Mapping the Antibody Response¶
For the final exercise, we will visualize the affinity of the antibody response to different regions of the HIV envelope protein. The data come from experiments in which animals are given an HIV vaccine, and pre- and post-vaccination samples are tested on an HIV peptide array containing thousands of HIV envelope sequences. Each array has triplicates of each sequence.
We need to do the following for each animal:
- read in pre-vaccination peptide array data a text file
- read in post-vaccination peptide array data a text file
- find the change in fluorescence reading from pre- to post-vaccination sample
- find the median value across the 3 replicates per sequence
- extract the clade data for each peptide
- read in a metadata file showing the mapping of peptide sequence to a region on the HIV envelope
- find the region each sequence is mapped to
Then, the investigator has asked us to plot the following heatmap where each cell contains the sum of values for sequences belonging to that clade, normalized by the number of such sequences in each clade.
heatmap
This non-trivial exercise will clearly test your mastery of the workshop data manipulation and visualization lessons!
The data comes from the paper Schiffner et al., J Virol. 2013 Sep;87(18):10163-72.
In [1]:
import os
import glob
import numpy as np
import pandas as pd
from pandas import DataFrame, Series
import matplotlib.pyplot as plt
import seaborn as sns
%matplotlib inline
Preview¶
Here is some code that can generate the above figure from raw data, but it is likely to be incomprehensible. Later we will unpack the code into the individual steps.
Paths to data and metadata files¶
We use os.path.join to create paths to files that will work
regardless of the operating system. In particular, Windows uses the
forward slash while Unix and Macs use the backwards slash to separate
parts of the path.
In [2]:
base_dir = os.path.join('data', 'capstone2')
meta_path = os.path.join(base_dir, 'Copy_Peptides Lib_HIV_Revised BK_20160905.txt')
pre_files = sorted(glob.glob(os.path.join(base_dir, '*pre_*anon.txt')))
post_files = sorted(glob.glob(os.path.join(base_dir, '*wk24_*anon.txt')))
pairs = list(zip(pre_files, post_files))
Step-by-step¶
What are the paths to files that were created?¶
In [3]:
base_dir
Out[3]:
'data/capstone2'
In [4]:
meta_path
Out[4]:
'data/capstone2/Copy_Peptides Lib_HIV_Revised BK_20160905.txt'
In [5]:
pairs
Out[5]:
[('data/capstone2/770 pre_20130118_S1_460_anon.txt',
'data/capstone2/770 wk24_20130118_S2_460_anon.txt'),
('data/capstone2/771 pre_20130118_S3_460_anon.txt',
'data/capstone2/771 wk24_20130118_S4_460_anon.txt'),
('data/capstone2/776 pre_20130118_S5_460_anon.txt',
'data/capstone2/776 wk24_20130118_S6_460_anon.txt'),
('data/capstone2/778 pre_20130118_S7_460_anon.txt',
'data/capstone2/778 wk24_20130118_S8_460_anon.txt'),
('data/capstone2/780 pre_20130118_S9_460_anon.txt',
'data/capstone2/780 wk24_20130118_S10_460_anon.txt')]
Load the metadata file into a pandas data frame¶
Looking at the original text file, we see that - indicates blank
space and can be coded as missing. Also we note that there are blank
rows in the text file that we must take care of. We take a peek at the
text file using Unix shell commands (the ! sends commands to the
shell).
In [6]:
! head -n 15 "$meta_path"
Use pandas to read in table¶
In [7]:
meta = pd.read_table(meta_path, na_values='-')
We have some blank rows¶
In [8]:
meta.ix[:11, :8]
Out[8]:
| OriNum | NewNum | PeptideSeq | Region | Seqs | N.ident | Batch | 12 | |
|---|---|---|---|---|---|---|---|---|
| 0 | 1.0 | E1 | MRVMGIQRNWQHLWR | 1.0 | 1 | 1.0 | Ori | NaN |
| 1 | 2.0 | E2 | MRVMGIQRNCQHLWR | 1.0 | 2 | 1.0 | Ori | NaN |
| 2 | 3.0 | E3 | MRVKGIRKNYQHLWR | 1.0 | 3 | 1.0 | Ori | NaN |
| 3 | 4.0 | E4 | MRVRGILRNWQQWWI | 1.0 | 4 | 1.0 | Ori | NaN |
| 4 | 5.0 | E5 | MRVRGIERNYQHLWR | 1.0 | 5 | 1.0 | Ori | NaN |
| 5 | 6.0 | E6 | MRVKETQMNWPNLWK | 1.0 | 6 | 1.0 | Ori | NaN |
| 6 | 7.0 | E7 | MRVMGIQKNYPLLWR | 1.0 | 7 | 1.0 | Ori | NaN |
| 7 | NaN | E1426 | MRVRGIWKNWPQWLI | 1.0 | 11 | 1.0 | New | NaN |
| 8 | NaN | E1427 | MRVMGTQKNCQQWWI | 1.0 | 12 | 1.0 | New | NaN |
| 9 | NaN | E1920 | MRVREILRNWQRWWT | 1.0 | 13 | 1.0 | New | C.ZM651 |
| 10 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 11 | 8.0 | E8 | MGIQRNWQHLWRWGT | 2.0 | 1 | 1.0 | Ori | NaN |
Now get rid of blank rows¶
In [9]:
meta = meta.dropna(axis=0, how='all')
Check that blank rows have been removed¶
In [10]:
meta.ix[:11, :8]
Out[10]:
| OriNum | NewNum | PeptideSeq | Region | Seqs | N.ident | Batch | 12 | |
|---|---|---|---|---|---|---|---|---|
| 0 | 1.0 | E1 | MRVMGIQRNWQHLWR | 1.0 | 1 | 1.0 | Ori | NaN |
| 1 | 2.0 | E2 | MRVMGIQRNCQHLWR | 1.0 | 2 | 1.0 | Ori | NaN |
| 2 | 3.0 | E3 | MRVKGIRKNYQHLWR | 1.0 | 3 | 1.0 | Ori | NaN |
| 3 | 4.0 | E4 | MRVRGILRNWQQWWI | 1.0 | 4 | 1.0 | Ori | NaN |
| 4 | 5.0 | E5 | MRVRGIERNYQHLWR | 1.0 | 5 | 1.0 | Ori | NaN |
| 5 | 6.0 | E6 | MRVKETQMNWPNLWK | 1.0 | 6 | 1.0 | Ori | NaN |
| 6 | 7.0 | E7 | MRVMGIQKNYPLLWR | 1.0 | 7 | 1.0 | Ori | NaN |
| 7 | NaN | E1426 | MRVRGIWKNWPQWLI | 1.0 | 11 | 1.0 | New | NaN |
| 8 | NaN | E1427 | MRVMGTQKNCQQWWI | 1.0 | 12 | 1.0 | New | NaN |
| 9 | NaN | E1920 | MRVREILRNWQRWWT | 1.0 | 13 | 1.0 | New | C.ZM651 |
| 11 | 8.0 | E8 | MGIQRNWQHLWRWGT | 2.0 | 1 | 1.0 | Ori | NaN |
Extract clades from meta¶
Each peptide sequence may match one or more HIV clades. The clade
information is in the columns
'12', '11', '10', '9', '8', '1', '2', '3', '4', '5', '6', '7', 'Unnamed: 19', 'Unnamed: 20',
and we will need to extract them from there.
In [11]:
clade_cols = ['12', '11', '10', '9', '8', '1', '2', '3', '4', '5', '6', '7', 'Unnamed: 19', 'Unnamed: 20']
meta[clade_cols].head()
Out[11]:
| 12 | 11 | 10 | 9 | 8 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | Unnamed: 19 | Unnamed: 20 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | NaN | NaN | NaN | NaN | NaN | NaN | M Con | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| 1 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | A Con | NaN | NaN | NaN | NaN | NaN | NaN |
| 2 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | B Con | NaN | NaN | NaN | NaN | NaN |
| 3 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | C Con | NaN | NaN | NaN | NaN |
| 4 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | D Con | NaN | NaN | AE.A244 |
First find all unique clades in the values within clade_cols¶
In [12]:
clades = sorted(set(meta[clade_cols].values.ravel()) - set([np.nan]))
In [13]:
clades
Out[13]:
['A Con',
'AE Con',
'AE.A244',
'AE.TH023',
'AG Con',
'B Con',
'B.MN',
'C Con',
'C.1086',
'C.TV1',
'C.ZM651',
'D Con',
'M Con']
Create a new column for each clade where the value is True if the clade is present in that row and false otherwise¶
In [14]:
for clade in clades:
meta[clade] = (meta[clade_cols] == clade).any(axis=1)
In [15]:
meta.columns
Out[15]:
Index(['OriNum', 'NewNum', 'PeptideSeq', 'Region', 'Seqs', 'N.ident', 'Batch',
'12', '11', '10', '9', '8', '1', '2', '3', '4', '5', '6', '7',
'Unnamed: 19', 'Unnamed: 20', 'A Con', 'AE Con', 'AE.A244', 'AE.TH023',
'AG Con', 'B Con', 'B.MN', 'C Con', 'C.1086', 'C.TV1', 'C.ZM651',
'D Con', 'M Con'],
dtype='object')
Drop all columns except Region, PeptideSeq and the columns for clades¶
In [16]:
meta = meta[['PeptideSeq', 'Region'] + clades]
meta.head()
Out[16]:
| PeptideSeq | Region | A Con | AE Con | AE.A244 | AE.TH023 | AG Con | B Con | B.MN | C Con | C.1086 | C.TV1 | C.ZM651 | D Con | M Con | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | MRVMGIQRNWQHLWR | 1.0 | False | False | False | False | False | False | False | False | False | False | False | False | True |
| 1 | MRVMGIQRNCQHLWR | 1.0 | True | False | False | False | False | False | False | False | False | False | False | False | False |
| 2 | MRVKGIRKNYQHLWR | 1.0 | False | False | False | False | False | True | False | False | False | False | False | False | False |
| 3 | MRVRGILRNWQQWWI | 1.0 | False | False | False | False | False | False | False | True | False | False | False | False | False |
| 4 | MRVRGIERNYQHLWR | 1.0 | False | False | True | False | False | False | False | False | False | False | False | True | False |
Reshape meta so that it only contains 4 columns PeptideSeq, Region , variable and value¶
Then drop all rows where the value of variable is False.
In [17]:
meta = pd.melt(meta, ['PeptideSeq', 'Region'])
meta.head()
Out[17]:
| PeptideSeq | Region | variable | value | |
|---|---|---|---|---|
| 0 | MRVMGIQRNWQHLWR | 1.0 | A Con | False |
| 1 | MRVMGIQRNCQHLWR | 1.0 | A Con | True |
| 2 | MRVKGIRKNYQHLWR | 1.0 | A Con | False |
| 3 | MRVRGILRNWQQWWI | 1.0 | A Con | False |
| 4 | MRVRGIERNYQHLWR | 1.0 | A Con | False |
In [18]:
meta = meta[meta.value == True].drop('value', axis=1)
meta.head()
Out[18]:
| PeptideSeq | Region | variable | |
|---|---|---|---|
| 1 | MRVMGIQRNCQHLWR | 1.0 | A Con |
| 11 | MGIQRNCQHLWRWGT | 2.0 | A Con |
| 21 | QRNCQHLWRWGTMIL | 3.0 | A Con |
| 31 | CQHLWRWGTMILGMI | 4.0 | A Con |
| 40 | LWRWGTMILGMIIIC | 5.0 | A Con |
Reanme the columns of meta as seq, region, clade¶
In [19]:
meta.columns = ['seq', 'region', 'clade']
Load a data file into a dataframe¶
We’ll just use one of the pre files as an example.
In [20]:
path = pairs[0][0]
path
Out[20]:
'data/capstone2/770 pre_20130118_S1_460_anon.txt'
In [21]:
! head -n 15 "$path"
ATF 1.0
32 58
"Type=GenePix Export 3"
"DateTime=2013/01/18 16:53:39"
"Settings="
"GalFile="
"PixelSize=10"
"Wavelengths=635 532"
"ImageFiles="
"NormalizationMethod=None"
"NormalizationFactors=1 1"
"JpegImage="
"StdDev=Type 1"
"RatioFormulations=W1/W2 (635/532)"
"FeatureType=Circular"
Hmm, the top part of the file does not actually contain the data we are looking for¶
It turns out that the first 34 lines are information about the assay we don’t need for this exercise. We use some Unix shell commands to show the lines 31-40.
In [22]:
! head -n 40 "$path" | tail -n 10
"ScanArea=1"
"Supplier=Arrayjet Ltd"
"ArrayerSoftwareName=Aj Marathon"
"ArrayerSoftwareVersion=1.3.4"
"Flags" "Normalize" "Autoflag" "Block" "Column" "Row" "Name" "ID" "X" "Y" "Dia." "F635 Median" "F635 Mean" "F635 SD" "F635 CV" "B635" "B635 Median" "B635 Mean" "B635 SD" "B635 CV" "% > B635+1SD" "% > B635+2SD" "F635 % Sat." "F532 Median" "F532 Mean" "F532 SD" "F532 CV" "B532" "B532 Median" "B532 Mean" "B532 SD" "B532 CV" "% > B532+1SD" "% > B532+2SD" "F532 % Sat." "F635 Total Intensity" "F532 Total Intensity" "SNR 635" "SNR 532" "Ratio of Medians (635/532)" "Ratio of Means (635/532)" "Median of Ratios (635/532)" "Mean of Ratios (635/532)" "Ratios SD (635/532)" "Rgn Ratio (635/532)" "Rgn R2 (635/532)" "Log Ratio (635/532)" "F635 Median - B635" "F532 Median - B532" "F635 Mean - B635" "F532 Mean - B532" "Sum of Medians (635/532)" "Sum of Means (635/532)" "F Pixels" "B Pixels" "Circularity" "Index" "Annotation"
0 0 0 1 1 1 "E73-73" "WVTVYYGVPVWKDAE" 2050 5160 100 347 359 160 44 267 267 276 84 30 49 6 0 2844 2962 1104 37 605 605 681 431 63 98 93 0 28686 236979 0.988 5.292 0.036 0.039 0.040 0.038 2.916 0.050 0.241 -4.807 80 2239 92 2357 2319 2449 80 560 100 1 "2729_1_2|798770|10010268_A09_1014|30006056_E09_0650
"
0 0 0 1 2 1 "E8-8" "MGIQRNWQHLWRWGT" 2350 5170 60 602 588 75 12 292 292 292 60 20 100 97 0 1283 1199 448 37 551 551 627 390 62 69 44 0 18824 38372 4.933 1.467 0.423 0.457 0.394 0.459 2.407 0.273 0.331 -1.240 310 732 296 648 1042 944 32 176 100 2 "2729_1_1|798676|10010267_A11_1103|30006056_A11_0861
"
-50 0 0 1 3 1 "E1932" "FCASDAKSYEKEVHN" 2640 5180 140 142 156 64 41 327 327 332 79 23 0 0 0 641 734 518 70 615 615 1051 3998 380 0 0 0 24295 114537 -2.228 -0.079 -7.115 -1.437 0.618 0.758 2.609 -0.001 0.000 Error -185 26 -171 119 -159 -52 156 1140 100 3 "2729_1_2|798820|10010268_E11_1219|30006056_G11_0959
Use pandas to read in the file, dropping the first 34 rows¶
In [23]:
pre = pd.read_table(path, skiprows=34)
In [24]:
pre.ix[:10, :8]
Out[24]:
| Flags | Normalize | Autoflag | Block | Column | Row | Name | ID | |
|---|---|---|---|---|---|---|---|---|
| 0 | 0 | 0 | 0 | 1 | 1 | 1 | E73-73 | WVTVYYGVPVWKDAE |
| 1 | 0 | 0 | 0 | 1 | 2 | 1 | E8-8 | MGIQRNWQHLWRWGT |
| 2 | -50 | 0 | 0 | 1 | 3 | 1 | E1932 | FCASDAKSYEKEVHN |
| 3 | 0 | 0 | 0 | 1 | 4 | 1 | E1435 | WLIWSILGFWIGNME |
| 4 | 0 | 0 | 0 | 1 | 5 | 1 | E1449 | EDLWVTVYYGVPVWR |
| 5 | 0 | 0 | 0 | 1 | 6 | 1 | E1427 | MRVMGTQKNCQQWWI |
| 6 | -50 | 0 | 0 | 1 | 7 | 1 | E1467 | FCASDAKAQETEAHN |
| 7 | -50 | 0 | 0 | 1 | 8 | 1 | E42-42 | MILGMIIICSAAENL |
| 8 | 0 | 0 | 0 | 1 | 9 | 1 | E75-75 | WVTVYYGVPVWRDAD |
| 9 | 0 | 0 | 0 | 1 | 10 | 1 | E7-7 | MRVMGIQKNYPLLWR |
| 10 | 0 | 0 | 0 | 1 | 11 | 1 | E105-105 | FCASDAKSYKTEAHN |
Drop unused columns¶
For this analysis, we are only interested in the columns named “ID”,
“Block” and “F635 Median - B635”, and we can drop the rest of
the many columns for simplicity. We never, ever touch the raw data
files, so we can always get the other columns by re-reading them in if
necessary.
In [25]:
pre = pre[['ID', 'Block', 'F635 Median - B635']]
In [26]:
pre.head()
Out[26]:
| ID | Block | F635 Median - B635 | |
|---|---|---|---|
| 0 | WVTVYYGVPVWKDAE | 1 | 80 |
| 1 | MGIQRNWQHLWRWGT | 1 | 310 |
| 2 | FCASDAKSYEKEVHN | 1 | -185 |
| 3 | WLIWSILGFWIGNME | 1 | 158 |
| 4 | EDLWVTVYYGVPVWR | 1 | 568 |
Do the same thing for the file in pairs[0][1] and create a post data frame. All the data files have the exact same format, so you must drop the first 34 rows as before.¶
In [27]:
path = pairs[0][1]
post = pd.read_table(path, skiprows=34)
post = post[['ID', 'Block', 'F635 Median - B635']]
In [28]:
post.head()
Out[28]:
| ID | Block | F635 Median - B635 | |
|---|---|---|---|
| 0 | WVTVYYGVPVWKDAE | 1 | 0 |
| 1 | MGIQRNWQHLWRWGT | 1 | 82 |
| 2 | FCASDAKSYEKEVHN | 1 | -13 |
| 3 | WLIWSILGFWIGNME | 1 | 119 |
| 4 | EDLWVTVYYGVPVWR | 1 | 954 |
Find the change in intensity before and after vaccination¶
First change the column names for convenience¶
- For
pre, call the columnsseq,block,pre - For
postcall the columnsseq,block,post
In [29]:
pre.columns = ['seq', 'block', 'pre']
post.columns = ['seq', 'block', 'post']
Calculate summary statistics¶
- from
pre, create a new data framebaselinethat contains seq and a column equal to the median value across blocks + 3 SD - if the resulting value is less than 200, use 200 instead - for
postcreate a new data framevaccinefor the median value across blocks
In [30]:
def baseline(x):
return max(200, x.median() + 3*x.std(ddof=1))
In [31]:
base = pre.groupby('seq').agg({'pre': baseline})
base = base.reset_index()
base.head()
Out[31]:
| seq | pre | |
|---|---|---|
| 0 | AA | 200.000000 |
| 1 | AAARLGAVILFVVIV | 200.000000 |
| 2 | AARAVELLGRSSLRG | 335.874922 |
| 3 | AARIVELLGRRGWEA | 537.473794 |
| 4 | AARTVELLGHSSLKG | 200.000000 |
In [32]:
vaccine = post.groupby('seq').agg({'post': np.median})
vaccine = vaccine.reset_index()
vaccine.head()
Out[32]:
| seq | post | |
|---|---|---|
| 0 | AA | -94 |
| 1 | AAARLGAVILFVVIV | -54 |
| 2 | AARAVELLGRSSLRG | 492 |
| 3 | AARIVELLGRRGWEA | 636 |
| 4 | AARTVELLGHSSLKG | 357 |
Create a datta frame df by merging baseline and vaccine on seq¶
In [33]:
df = pd.merge(base, vaccine, on=['seq'])
df.head()
Out[33]:
| seq | pre | post | |
|---|---|---|---|
| 0 | AA | 200.000000 | -94 |
| 1 | AAARLGAVILFVVIV | 200.000000 | -54 |
| 2 | AARAVELLGRSSLRG | 335.874922 | 492 |
| 3 | AARIVELLGRRGWEA | 537.473794 | 636 |
| 4 | AARTVELLGHSSLKG | 200.000000 | 357 |
Create a new column signal in df by subtracting the pre column from the post column¶
If the resulting value is less than 1, use 1 instead.
In [34]:
df['signal'] = df['post'] - df['pre']
df['signal'] = df['signal'].clip_lower(1)
df.head()
Out[34]:
| seq | pre | post | signal | |
|---|---|---|---|---|
| 0 | AA | 200.000000 | -94 | 1.000000 |
| 1 | AAARLGAVILFVVIV | 200.000000 | -54 | 1.000000 |
| 2 | AARAVELLGRSSLRG | 335.874922 | 492 | 156.125078 |
| 3 | AARIVELLGRRGWEA | 537.473794 | 636 | 98.526206 |
| 4 | AARTVELLGHSSLKG | 200.000000 | 357 | 157.000000 |
Drop the pre and post columns since we don’t need them anymore¶
In [35]:
df = df.drop(['pre', 'post'], axis=1)
df.head()
Out[35]:
| seq | signal | |
|---|---|---|
| 0 | AA | 1.000000 |
| 1 | AAARLGAVILFVVIV | 1.000000 |
| 2 | AARAVELLGRSSLRG | 156.125078 |
| 3 | AARIVELLGRRGWEA | 98.526206 |
| 4 | AARTVELLGHSSLKG | 157.000000 |
Merge with metadta¶
Merge df and meta on seq to create a new data frame called data¶
In [36]:
data = pd.merge(df, meta, on='seq')
In [37]:
data.head()
Out[37]:
| seq | signal | region | clade | |
|---|---|---|---|---|
| 0 | AARAVELLGRSSLRG | 156.125078 | 254.0 | C Con |
| 1 | AARIVELLGRRGWEA | 98.526206 | 254.0 | D Con |
| 2 | AARTVELLGHSSLKG | 157.000000 | 254.0 | A Con |
| 3 | AARTVELLGHSSLKG | 157.000000 | 254.0 | AE Con |
| 4 | AARTVELLGHSSLKG | 157.000000 | 254.0 | AG Con |
Now do this for all files in a loop¶
Refactor code into functions to make the code more manageable¶
In [38]:
def process_meta(path, clade_cols):
"""Collect steps to transform metadata here."""
meta = pd.read_table(meta_path, na_values='-')
clades = sorted(set(meta[clade_cols].values.ravel()) - set([np.nan]))
for clade in clades:
meta[clade] = (meta[clade_cols] == clade).any(axis=1)
meta = meta[['PeptideSeq', 'Region'] + clades]
meta = pd.melt(meta, ['PeptideSeq', 'Region'])
meta = meta[meta.value == True].drop('value', axis=1)
meta.columns = ['seq', 'region', 'clade']
return meta
def process_baseline(path):
"""Steps to calculate baseline values."""
pre = pd.read_table(path, skiprows=34)
pre = pre[['ID', 'Block', 'F635 Median - B635']]
pre.columns = ['seq', 'block', 'pre']
d = pre.groupby('seq').agg({'pre': baseline})
d = d.reset_index()
return d
def process_vaccinated(path):
"""Steps to calculate vaccinated median values."""
post = pd.read_table(path, skiprows=34)
post = post[['ID', 'Block', 'F635 Median - B635']]
post.columns = ['seq', 'block', 'post']
d = post.groupby('seq').agg({'post': np.median})
d = d.reset_index()
return d
def process_pair(pre_path, post_path):
"""Process a pair of pre- and post-files."""
s1 = os.path.split(pre_path)[-1][:3]
s2 = os.path.split(pre_path)[-1][:3]
assert(s1 == s2)
pre = process_baseline(pre_path)
post = process_vaccinated(post_path)
df = pd.merge(pre, post, on=['seq'])
df['signal'] = df['post'] - df['pre']
df['signal'] = df['signal'].clip_lower(1)
df['subject'] = s1
df = df.drop(['pre', 'post'], axis=1)
return df
In [39]:
def process_all(meta_path, pairs, clade_cols):
"""Process metadata and all pairs of files into a single data frame."""
meta = meta = process_meta(meta_path, clade_cols)
dfs = []
for pre_path, post_path in pairs:
df = process_pair(pre_path, post_path)
dfs.append(df)
dfs = pd.concat(dfs)
data = pd.merge(dfs, meta)
return data
In [40]:
clade_cols = ['12', '11', '10', '9', '8', '1', '2', '3', '4', '5', '6', '7', 'Unnamed: 19', 'Unnamed: 20']
base_dir = os.path.join('data', 'capstone2')
meta_path = os.path.join(base_dir, 'Copy_Peptides Lib_HIV_Revised BK_20160905.txt')
pre_files = sorted(glob.glob(os.path.join(base_dir, '*pre_*anon.txt')))
post_files = sorted(glob.glob(os.path.join(base_dir, '*wk24_*anon.txt')))
pairs = list(zip(pre_files, post_files))
In [41]:
data = process_all(meta_path, pairs, clade_cols)
data.head()
Out[41]:
| seq | signal | subject | region | clade | |
|---|---|---|---|---|---|
| 0 | AARAVELLGRSSLRG | 156.125078 | 770 | 254.0 | C Con |
| 1 | AARAVELLGRSSLRG | 1.000000 | 771 | 254.0 | C Con |
| 2 | AARAVELLGRSSLRG | 1.000000 | 776 | 254.0 | C Con |
| 3 | AARAVELLGRSSLRG | 1.000000 | 778 | 254.0 | C Con |
| 4 | AARAVELLGRSSLRG | 1.000000 | 780 | 254.0 | C Con |
Assigned groups¶
Group information GLA-Rb Study:
- WT Group: 770, 771, 776
- GLA Group: 778, 780, 783
In [42]:
wt = ['770', '771', '776']
gla = ['778', '780', '783']
In [43]:
data.ix[data.subject.isin(wt), 'group'] = 'wt'
data.ix[data.subject.isin(gla), 'group'] = 'gla'
data.head()
Out[43]:
| seq | signal | subject | region | clade | group | |
|---|---|---|---|---|---|---|
| 0 | AARAVELLGRSSLRG | 156.125078 | 770 | 254.0 | C Con | wt |
| 1 | AARAVELLGRSSLRG | 1.000000 | 771 | 254.0 | C Con | wt |
| 2 | AARAVELLGRSSLRG | 1.000000 | 776 | 254.0 | C Con | wt |
| 3 | AARAVELLGRSSLRG | 1.000000 | 778 | 254.0 | C Con | gla |
| 4 | AARAVELLGRSSLRG | 1.000000 | 780 | 254.0 | C Con | gla |
Assigned envelope proteins¶
If region <= 163, then protein = gp120, otherwise protein = gp144
Graphics¶
In [44]:
data.ix[data.region <= 163, 'protein'] = 'gp120'
data.ix[data.region >163, 'protein'] = 'gp144'
In [45]:
data.head()
Out[45]:
| seq | signal | subject | region | clade | group | protein | |
|---|---|---|---|---|---|---|---|
| 0 | AARAVELLGRSSLRG | 156.125078 | 770 | 254.0 | C Con | wt | gp144 |
| 1 | AARAVELLGRSSLRG | 1.000000 | 771 | 254.0 | C Con | wt | gp144 |
| 2 | AARAVELLGRSSLRG | 1.000000 | 776 | 254.0 | C Con | wt | gp144 |
| 3 | AARAVELLGRSSLRG | 1.000000 | 778 | 254.0 | C Con | gla | gp144 |
| 4 | AARAVELLGRSSLRG | 1.000000 | 780 | 254.0 | C Con | gla | gp144 |
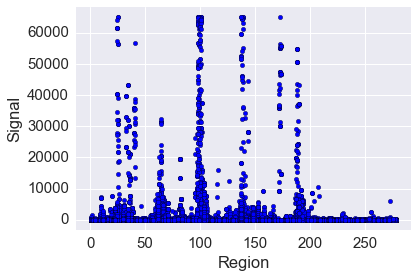
Plotting signal against region¶
In [46]:
plt.scatter(data.region, data.signal)
plt.margins(0.05)
plt.xlabel('Region')
plt.ylabel('Signal')
pass
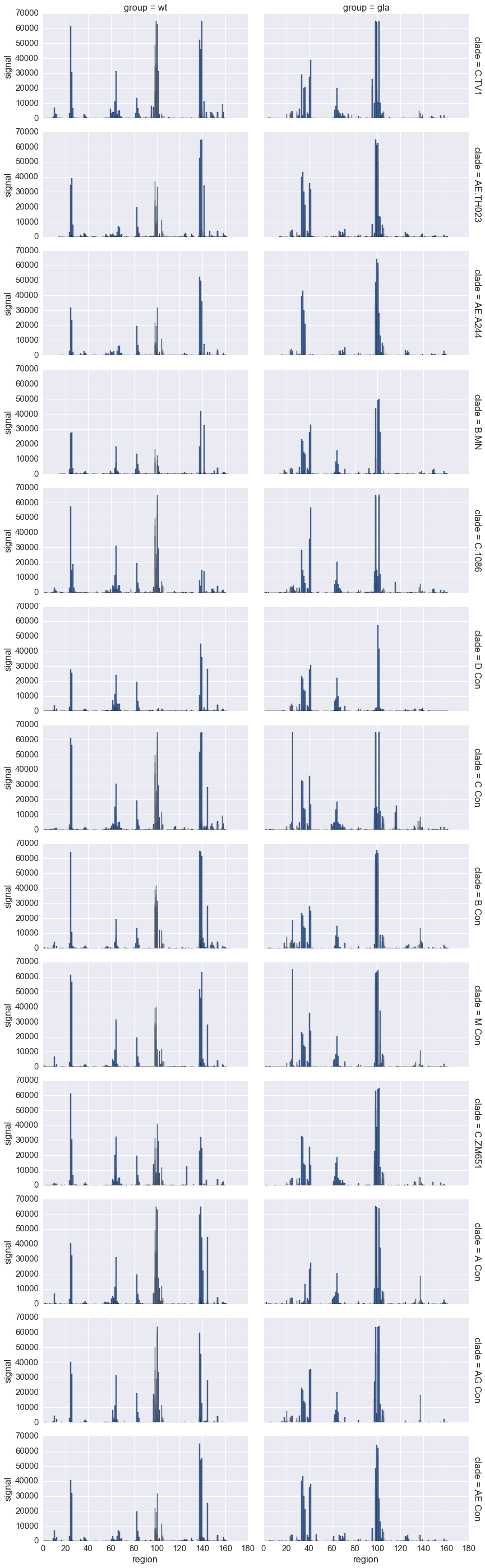
Conditioning plots on categorical variables¶
In [47]:
gp120 = data[data.protein == 'gp120']
g = sns.FacetGrid(gp120, row='clade', col='group', aspect=2, margin_titles=True)
g.map(plt.bar, 'region', 'signal')
pass
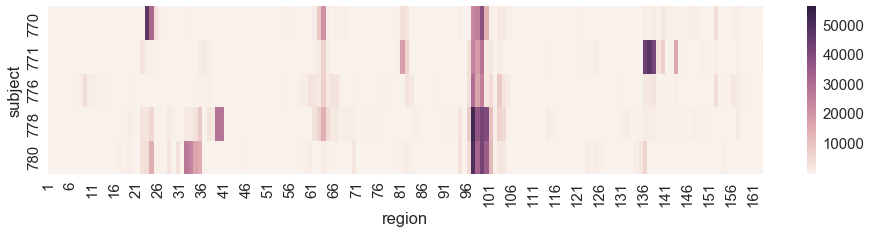
Plotting heatmaps¶
In [48]:
hm = data.groupby(['region', 'subject']).agg({'signal': np.mean})
hm.head()
Out[48]:
| signal | ||
|---|---|---|
| region | subject | |
| 1.0 | 770 | 1.000000 |
| 771 | 144.941271 | |
| 776 | 54.898099 | |
| 778 | 70.446511 | |
| 780 | 3.236776 |
In [49]:
hm = hm.unstack().T
hm.ix[:4, :6]
Out[49]:
| region | 1.0 | 2.0 | 3.0 | 4.0 | 5.0 | 6.0 | |
|---|---|---|---|---|---|---|---|
| subject | |||||||
| signal | 770 | 1.000000 | 65.258850 | 2.400000 | 1.000000 | 22.888889 | 30.400000 |
| 771 | 144.941271 | 99.295281 | 30.186508 | 94.206611 | 43.111111 | 24.425327 | |
| 776 | 54.898099 | 17.535661 | 1.000000 | 33.542959 | 38.111111 | 48.678128 | |
| 778 | 70.446511 | 111.469690 | 37.500000 | 135.000000 | 166.111111 | 126.300000 |
In [50]:
hm = hm.reset_index(level=0, drop=True)
hm.ix[:4, :6]
Out[50]:
| region | 1.0 | 2.0 | 3.0 | 4.0 | 5.0 | 6.0 |
|---|---|---|---|---|---|---|
| subject | ||||||
| 770 | 1.000000 | 65.258850 | 2.400000 | 1.000000 | 22.888889 | 30.400000 |
| 771 | 144.941271 | 99.295281 | 30.186508 | 94.206611 | 43.111111 | 24.425327 |
| 776 | 54.898099 | 17.535661 | 1.000000 | 33.542959 | 38.111111 | 48.678128 |
| 778 | 70.446511 | 111.469690 | 37.500000 | 135.000000 | 166.111111 | 126.300000 |
In [51]:
plt.figure(figsize=(16, 3))
sns.heatmap(hm)
xlim = [1, 163]
plt.xlim(xlim)
plt.xticks(range(xlim[0], xlim[1], 5), range(xlim[0], xlim[1], 5))
pass
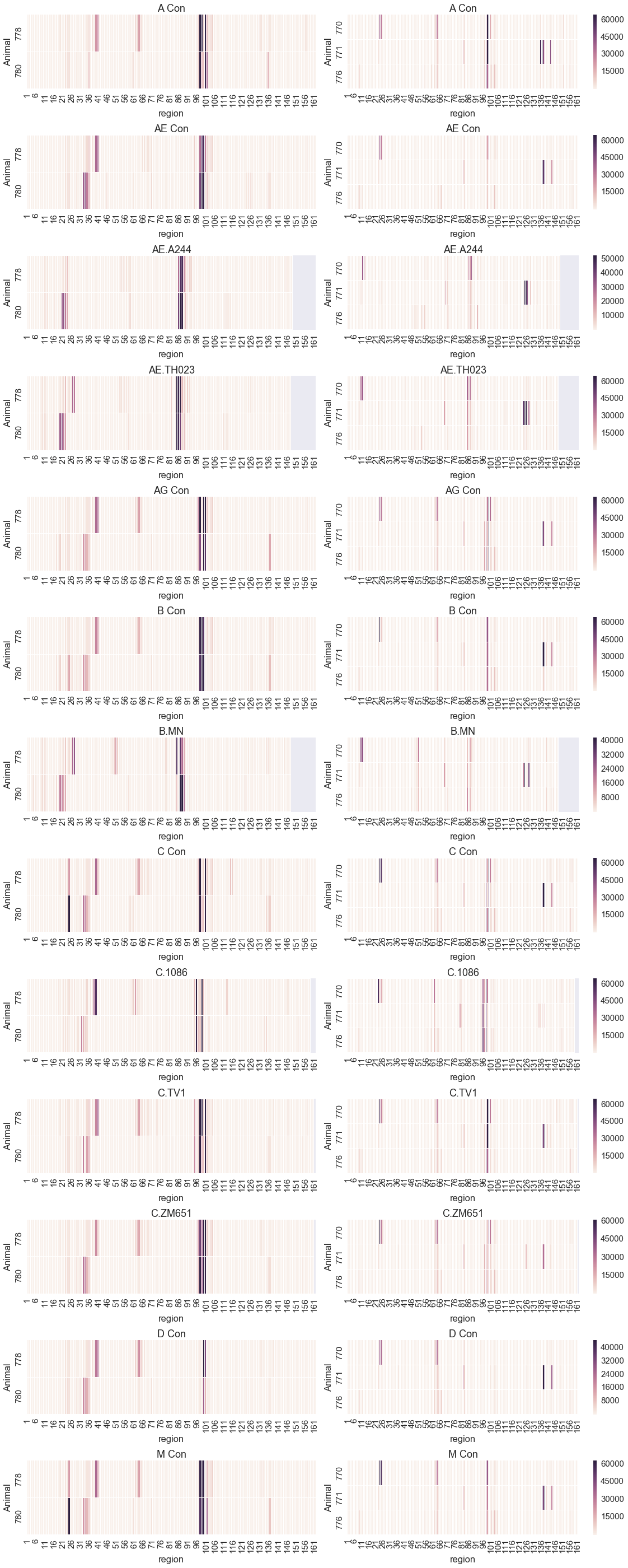
Conditioning heatmaps¶
In [52]:
nrows = len(clades)
ncols = 2
fig, axes = plt.subplots(nrows, ncols, figsize=(16, 3*nrows))
low, high, step = 1, 162, 5
for i, (clade, df_clade) in enumerate(data[data.protein == 'gp120'].groupby('clade')):
for j, (group, df_group) in enumerate(df_clade.groupby('group')):
ax = axes[i, j]
hm = df_group.groupby(['region', 'subject'])[['signal']].mean()
hm = hm.unstack().T.reset_index(level=0, drop=True)
hm.columns = hm.columns.astype('int')
sns.heatmap(hm, ax=ax, linewidth=0.01, cbar=j)
ax.set_title(clade)
ax.set_xlim(low, high)
ax.set_xticks(range(low, high+1, step))
ax.set_xticklabels(range(low, high+1, step))
ax.set_ylabel('Animal')
plt.tight_layout()
Code to remove identifying laboratory information¶
for f in glob.glob(os.path.join(base_dir, '7*txt')):
if 'anon' in f:
continue
with open(f) as fin:
with open(f.replace('.txt', '_anon.txt'), 'w') as fout:
for line in fin:
if line.startswith('"GalFile='):
fout.write('"GalFile="\n')
elif line.startswith('"ImageFiles='):
fout.write('"ImageFiles="\n')
else:
fout.write(line)