DESeq2 Analysis of 2015 Data¶
In this notebook, we will reproduce the dendrograms from the 2015 data shown in the welcome slides
Load packages¶
Load requisite R packages.
In [1]:
library(DESeq2)
library(tools)
library(dendextend)
library(dplyr)
library(RColorBrewer)
options(width=100)
Loading required package: S4Vectors
Loading required package: stats4
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:parallel’:
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from ‘package:stats’:
IQR, mad, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, append, as.data.frame, cbind, colnames, do.call,
duplicated, eval, evalq, Filter, Find, get, grep, grepl, intersect,
is.unsorted, lapply, lengths, Map, mapply, match, mget, order,
paste, pmax, pmax.int, pmin, pmin.int, Position, rank, rbind,
Reduce, rownames, sapply, setdiff, sort, table, tapply, union,
unique, unsplit, which, which.max, which.min
Attaching package: ‘S4Vectors’
The following objects are masked from ‘package:base’:
colMeans, colSums, expand.grid, rowMeans, rowSums
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: GenomeInfoDb
Loading required package: SummarizedExperiment
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
---------------------
Welcome to dendextend version 1.5.2
Type citation('dendextend') for how to cite the package.
Type browseVignettes(package = 'dendextend') for the package vignette.
The github page is: https://github.com/talgalili/dendextend/
Suggestions and bug-reports can be submitted at: https://github.com/talgalili/dendextend/issues
Or contact: <tal.galili@gmail.com>
To suppress this message use: suppressPackageStartupMessages(library(dendextend))
---------------------
Attaching package: ‘dendextend’
The following object is masked from ‘package:stats’:
cutree
Attaching package: ‘dplyr’
The following object is masked from ‘package:Biobase’:
combine
The following objects are masked from ‘package:GenomicRanges’:
intersect, setdiff, union
The following object is masked from ‘package:GenomeInfoDb’:
intersect
The following objects are masked from ‘package:IRanges’:
collapse, desc, intersect, regroup, setdiff, slice, union
The following objects are masked from ‘package:S4Vectors’:
first, intersect, rename, setdiff, setequal, union
The following objects are masked from ‘package:BiocGenerics’:
combine, intersect, setdiff, union
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
Prepare for Data Import¶
First set the directory under which the HTSeq count files are stored
In [2]:
datadir<-"~/work/HTS_SummerCourse_2017/Materials/Statistics/08032017/Data/2015"
Next, put the filenames into a data frame
In [3]:
phdata<-data.frame(fname=list.files(path=datadir,pattern="*.csv"),stringsAsFactors=FALSE)
It is always a good idea to check the dimension of the file you have read in
In [4]:
dim(phdata)
- 30
- 1
Extract the label from the filename.
In [5]:
phdata <- phdata %>% transmute(sample=substr(fname,1,4),fname)
Add some design info to the data frame. We will add the treatment factor (the first character of the sample string: 7 or 8), the replicate id (the second character in the
Note that tools::md5sum will add the MD5 signature for each of the HTSeq count files. You should keep track of these for the purpose of conducting reproducible analysis
In [6]:
phdata <- phdata %>%
mutate(trt=as.factor(substr(sample,1,1)),
repl=substr(sample,2,2),
team=substr(sample,4,4),
md5=tools::md5sum(file.path(datadir,fname)))
head(phdata)
| sample | fname | trt | repl | team | md5 | |
|---|---|---|---|---|---|---|
| 1 | 7A_E | 7A_E.csv | 7 | A | E | NA |
| 2 | 7A_G | 7A_G.csv | 7 | A | G | NA |
| 3 | 7A_K | 7A_K.csv | 7 | A | K | NA |
| 4 | 7A_N | 7A_N.csv | 7 | A | N | NA |
| 5 | 7A_P | 7A_P.csv | 7 | A | P | NA |
| 6 | 7B_E | 7B_E.csv | 7 | B | E | NA |
Now, we import the counts. Note that the first argument is the sample table while the second is the directory storing the count files. The last argument specifies the design. More on this later.
In [7]:
dds<-DESeqDataSetFromHTSeqCount(sampleTable=phdata,directory=datadir,design=~ trt)
Estimate library specific size factors and gene specific dispersion paramaters¶
Estimate Size factors
In [8]:
dds<-estimateSizeFactors(dds)
Estimate dispersion parameters
In [9]:
dds<-estimateDispersions(dds)
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
Inspect object
In [10]:
dds
class: DESeqDataSet
dim: 4444 30
metadata(1): version
assays(2): counts mu
rownames(4444): gene0 gene1 ... gene998 gene999
rowData names(9): baseMean baseVar ... dispOutlier dispMAP
colnames(30): 7A_E 7A_G ... 8C_N 8C_P
colData names(5): trt repl team md5 sizeFactor
Create dendrograms¶
For this tutorial, we will use t he expression data based on rlog transformation.
In [11]:
rlogexp<-assay(rlog(dds))
Next, we create the dendrogram object using hierarchical clustering using complete linkage (method=”complete”).
Note that you have you transpose the expression matrix as R assumes that the samples are along the rows and that the genes are across the columns. This is accomplished using the t() function. Next, you create the distance matrix for this expression matrix using the dist() function. Afterwards, you conduct hierachical clustering on the basis of complete linkage using this distance matrix usin the dist() function. Finally, the results from the clustering is converted into a dendrogram class using the as.dendrogram() function
In [13]:
dend <- rlogexp %>%
t %>%
dist %>%
hclust(method="complete") %>%
as.dendrogram
Let’s look as a plot
In [14]:
dend %>% plot
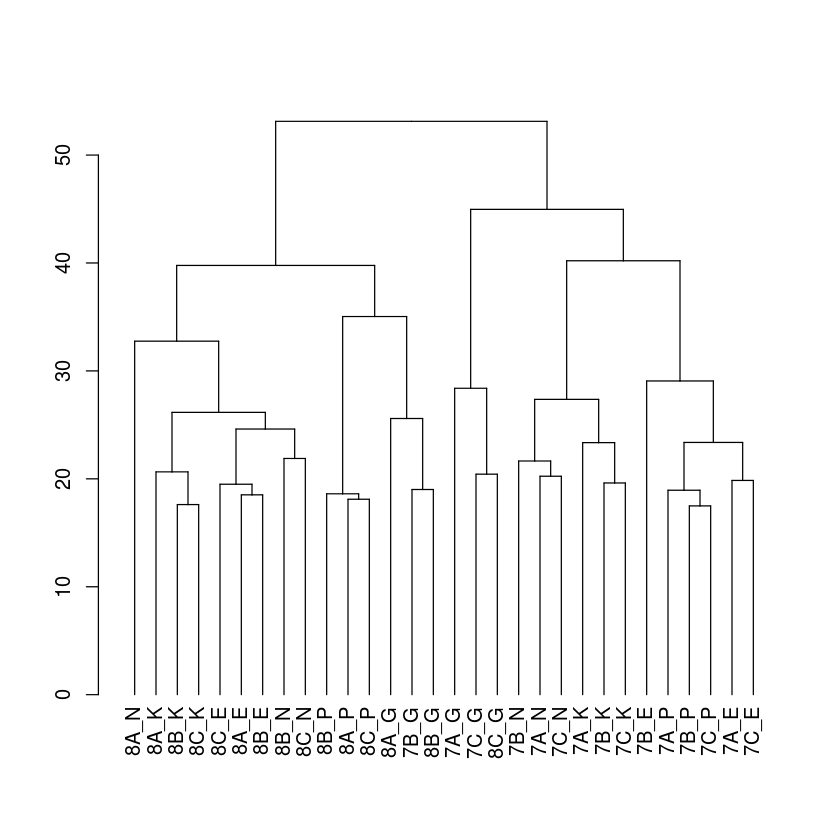
Next, we will add some eye candy to this dendrogram. We will add the treatment label as symbols and the group/team labels as colors. Note that one can extract the labels from the dendrogram using the labels() function
In [15]:
labels(dend)
- '8A_N'
- '8A_K'
- '8B_K'
- '8C_K'
- '8C_E'
- '8A_E'
- '8B_E'
- '8B_N'
- '8C_N'
- '8B_P'
- '8A_P'
- '8C_P'
- '8A_G'
- '7B_G'
- '8B_G'
- '7A_G'
- '7C_G'
- '8C_G'
- '7B_N'
- '7A_N'
- '7C_N'
- '7A_K'
- '7B_K'
- '7C_K'
- '7B_E'
- '7A_P'
- '7B_P'
- '7C_P'
- '7A_E'
- '7C_E'
Now use this information to create a treatment label vector
In [16]:
trtlab<-c(2,19)[factor(substr(labels(dend),1,1))]
trtlab
- 19
- 19
- 19
- 19
- 19
- 19
- 19
- 19
- 19
- 19
- 19
- 19
- 19
- 2
- 19
- 2
- 2
- 19
- 2
- 2
- 2
- 2
- 2
- 2
- 2
- 2
- 2
- 2
- 2
- 2
and then a group label vector
In [17]:
grplab<-brewer.pal(5,"Set1")[factor(substr(labels(dend),4,4))]
grplab
- '#984EA3'
- '#4DAF4A'
- '#4DAF4A'
- '#4DAF4A'
- '#E41A1C'
- '#E41A1C'
- '#E41A1C'
- '#984EA3'
- '#984EA3'
- '#FF7F00'
- '#FF7F00'
- '#FF7F00'
- '#377EB8'
- '#377EB8'
- '#377EB8'
- '#377EB8'
- '#377EB8'
- '#377EB8'
- '#984EA3'
- '#984EA3'
- '#984EA3'
- '#4DAF4A'
- '#4DAF4A'
- '#4DAF4A'
- '#E41A1C'
- '#FF7F00'
- '#FF7F00'
- '#FF7F00'
- '#E41A1C'
- '#E41A1C'
First add the treatment label. The symbol use for each leaf is controlled by “leaves_pch” (you can use label_cex and leaves_cex to control the relative size of the labels or leaves)
In [18]:
dend %>%
set("labels_cex",1) %>%
set("leaves_pch",trtlab) %>%
set("leaves_cex",1) %>%
plot
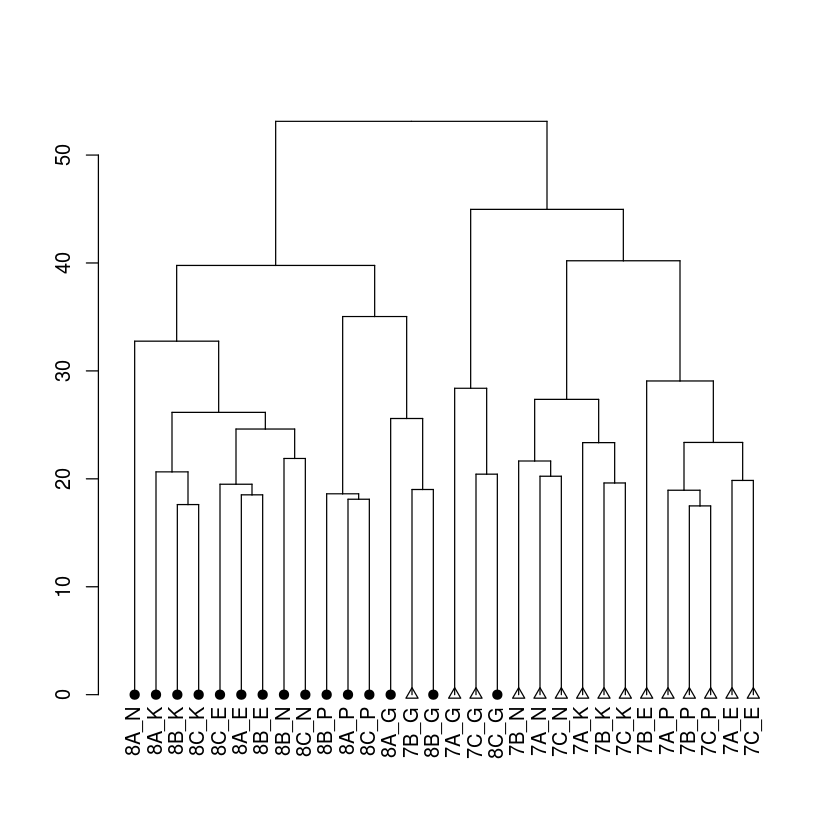
Now, lets add the group label to the above. The color of each label is controlled by “labels_col”
In [19]:
dend %>%
set("labels_cex",1) %>%
set("labels_col",grplab) %>%
set("leaves_pch",trtlab) %>%
set("leaves_cex",1) %>%
plot
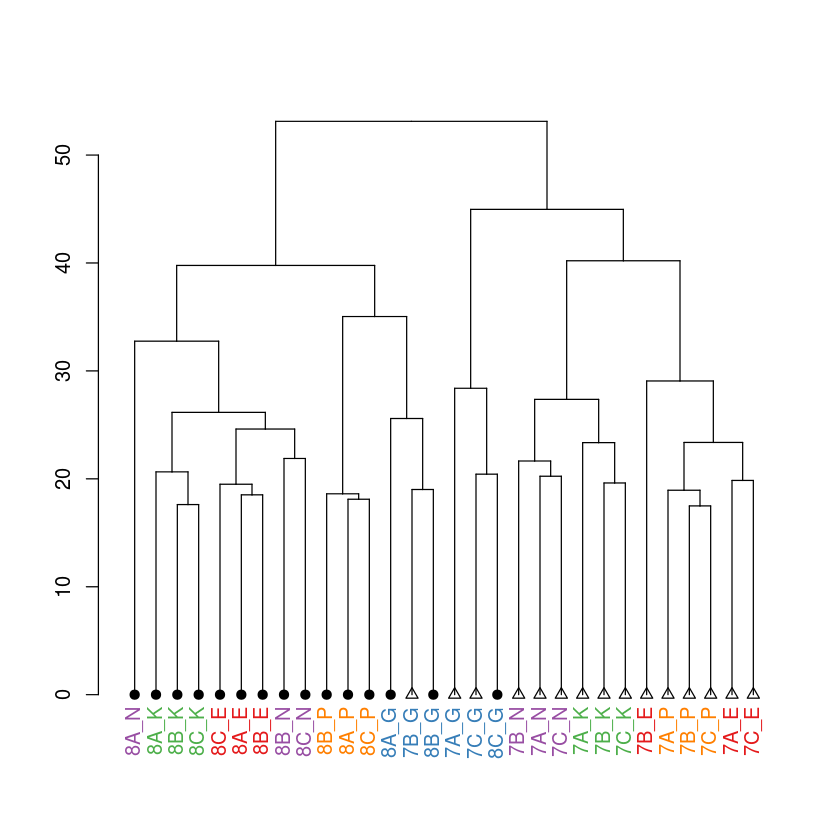
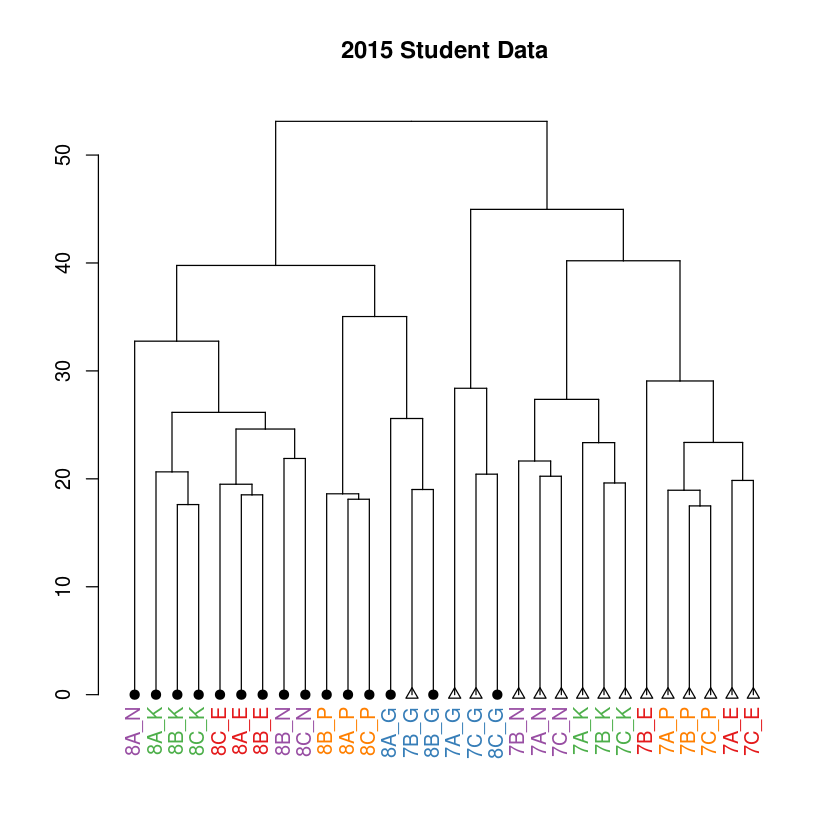
You can also add a title to the plot
In [20]:
dend %>%
set("labels_cex",1) %>%
set("labels_col",grplab) %>%
set("leaves_pch",trtlab) %>%
set("leaves_cex",1) %>%
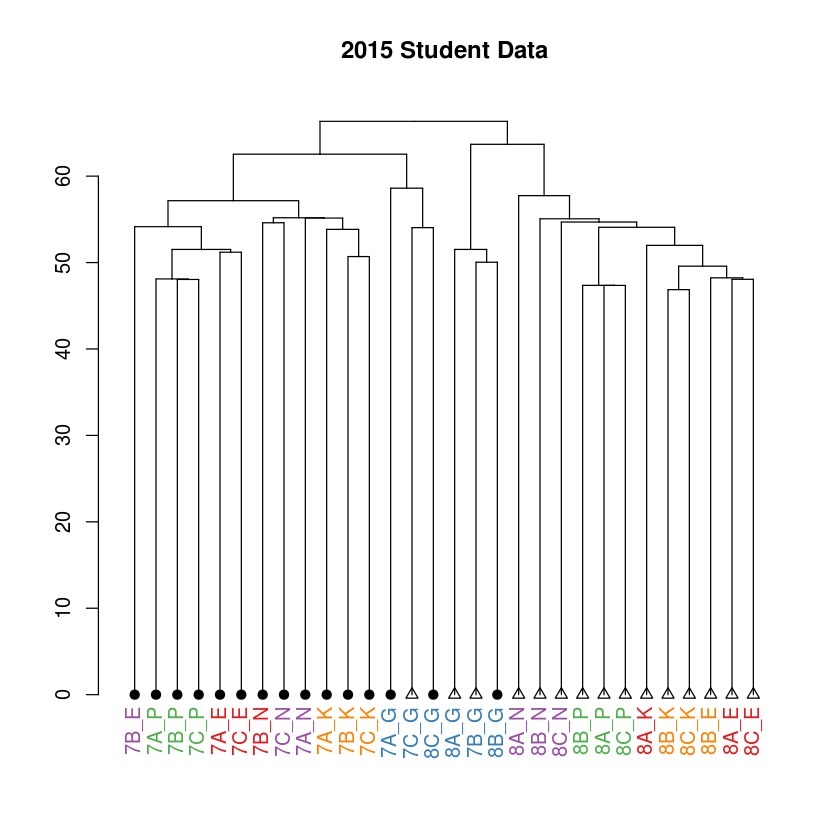
plot(main="2015 Student Data")
Repeat the analysis using expression data based on limma::voom with HC with single linkage:¶
Get expression matrix using voom
In [21]:
voomexp<-limma::voom(assay(dds))$E
Create dendrogram object based on single linkage
In [22]:
dend <- voomexp %>%
t %>%
dist %>%
hclust(method="single") %>%
as.dendrogram
Create plot
In [23]:
dend %>%
set("labels_cex",1) %>%
set("labels_col",grplab) %>%
set("leaves_pch",trtlab) %>%
set("leaves_cex",1) %>%
plot(main="2015 Student Data")
Get Session Information¶
In [24]:
sessionInfo()
R version 3.3.1 (2016-06-21)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Debian GNU/Linux 8 (jessie)
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8
[4] LC_COLLATE=en_US.UTF-8 LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] tools parallel stats4 stats graphics grDevices utils datasets methods
[10] base
other attached packages:
[1] RColorBrewer_1.1-2 dplyr_0.5.0 dendextend_1.5.2
[4] DESeq2_1.14.1 SummarizedExperiment_1.4.0 Biobase_2.34.0
[7] GenomicRanges_1.26.4 GenomeInfoDb_1.10.3 IRanges_2.8.2
[10] S4Vectors_0.12.2 BiocGenerics_0.20.0
loaded via a namespace (and not attached):
[1] viridis_0.4.0 viridisLite_0.2.0 jsonlite_1.1 splines_3.3.1
[5] Formula_1.2-2 assertthat_0.1 latticeExtra_0.6-28 robustbase_0.92-7
[9] RSQLite_1.0.0 backports_1.1.0 lattice_0.20-34 limma_3.30.13
[13] uuid_0.1-2 digest_0.6.10 XVector_0.14.1 checkmate_1.8.3
[17] colorspace_1.3-1 htmltools_0.3.5 Matrix_1.2-7.1 plyr_1.8.4
[21] XML_3.98-1.9 genefilter_1.56.0 zlibbioc_1.20.0 mvtnorm_1.0-6
[25] xtable_1.8-2 scales_0.4.1 whisker_0.3-2 BiocParallel_1.8.2
[29] htmlTable_1.9 tibble_1.2 annotate_1.52.1 ggplot2_2.2.1
[33] repr_0.7 nnet_7.3-12 lazyeval_0.2.0 survival_2.41-3
[37] magrittr_1.5 crayon_1.3.1 mclust_5.3 memoise_1.0.0
[41] evaluate_0.10 MASS_7.3-45 class_7.3-14 foreign_0.8-67
[45] data.table_1.10.4 trimcluster_0.1-2 stringr_1.0.0 kernlab_0.9-25
[49] munsell_0.4.3 locfit_1.5-9.1 cluster_2.0.5 AnnotationDbi_1.36.2
[53] fpc_2.1-10 grid_3.3.1 RCurl_1.95-4.8 pbdZMQ_0.2-3
[57] IRkernel_0.7 htmlwidgets_0.9 bitops_1.0-6 base64enc_0.1-3
[61] gtable_0.2.0 flexmix_2.3-14 DBI_0.5-1 R6_2.2.0
[65] gridExtra_2.2.1 prabclus_2.2-6 knitr_1.15.1 Hmisc_4.0-3
[69] modeltools_0.2-21 stringi_1.1.2 IRdisplay_0.4.3 Rcpp_0.12.8
[73] geneplotter_1.52.0 rpart_4.1-10 acepack_1.4.1 DEoptimR_1.0-8
[77] diptest_0.75-7