Extended example
Suppose we have the following scenario:
A multi-center experiment is conducted at Duke and UNC.
- Duke has patients
Ann, Bob, Charle
- UNC has patients
David, Ed, Fiona
- Each patient has blood sampled at 2 time points
- The genomics core at each center measures 3 genes for each patient at
each time point
- The proteomics core at each center measures 3 proteins from each
patient at each time point
- Each center has additional information about patient demographics in
a different database
We have a ten files to work with:
duke_demographics.tsv
duke_genes_v1.tsv
duke_genes_v2.tsv
duke_proteins_v1.tsv
duke_proteins_v2.tsv
unc_demographics.tsv
unc_genes_v1.tsv
unc_genes_v2.tsv
unc_proteins_v1.tsv
unc_proteins_v2.tsv
Objective
We want to visualize the correlation between the gene and protein
expression changes from visit 1 to visit 2 for all patients in the
study. We also want to augment the data with demographic variables for
each patient.
- Assume that labs within an institution do not have batch effects
across visits
- Assume that labs at different institutions have batch affects and
must be normalized before comparison
Combine gene expression data from Duke and normalize to zero mean and unit standard deviation
Parsed with column specification:
cols(
pet = col_character(),
gene_A = col_double(),
gene_B = col_double(),
gene_C = col_double()
)
Parsed with column specification:
cols(
pet = col_character(),
gene_A = col_double(),
gene_B = col_double(),
gene_C = col_double()
)
| pet | gene_A | gene_B | gene_C |
|---|
| Ann | 91.09792 | 64.62166 | 124.4608 |
| Bob | 88.81115 | 78.24940 | 113.6644 |
| Charlie | 107.52933 | 70.52984 | 124.5951 |
| pet | gene_A | gene_B | gene_C |
|---|
| Ann | 108.67919 | 90.38630 | 137.5395 |
| Bob | 113.44700 | 101.52725 | 144.7763 |
| Charlie | 98.12047 | 97.70863 | 131.1964 |
| visit | pet | gene_A | gene_B | gene_C |
|---|
| 1 | Ann | 91.09792 | 64.62166 | 124.4608 |
| 1 | Bob | 88.81115 | 78.24940 | 113.6644 |
| 1 | Charlie | 107.52933 | 70.52984 | 124.5951 |
| 2 | Ann | 108.67919 | 90.38630 | 137.5395 |
| 2 | Bob | 113.44700 | 101.52725 | 144.7763 |
| 2 | Charlie | 98.12047 | 97.70863 | 131.1964 |
| visit | pet | gene_A | gene_B | gene_C |
|---|
| 1 | Ann | -1.0073042 | -1.2805845 | -0.4479685 |
| 1 | Bob | -1.2335142 | -0.3723877 | -1.4327190 |
| 1 | Charlie | 0.6181059 | -0.8868438 | -0.4357137 |
| 2 | Ann | 0.7318518 | 0.4364545 | 0.7449587 |
| 2 | Bob | 1.2034879 | 1.1789234 | 1.4050414 |
| 2 | Charlie | -0.3126272 | 0.9244382 | 0.1664011 |
Do the same for the other data sets
Duke protein expression
Parsed with column specification:
cols(
pet = col_character(),
prot_A = col_double(),
prot_B = col_double(),
prot_C = col_double()
)
Parsed with column specification:
cols(
pet = col_character(),
prot_A = col_double(),
prot_B = col_double(),
prot_C = col_double()
)
| visit | pet | prot_A | prot_B | prot_C |
|---|
| 1 | Ann | -0.4967020 | 0.9830005 | -1.37774584 |
| 1 | Bob | -1.0132844 | -1.1799912 | -1.01496776 |
| 1 | Charlie | -0.3455620 | -0.8561590 | 0.60497113 |
| 2 | Ann | -0.4191436 | 0.3772850 | 0.88392265 |
| 2 | Bob | 1.7870904 | -0.5388750 | -0.05597907 |
| 2 | Charlie | 0.4876017 | 1.2147397 | 0.95979888 |
UNC gene expression
Parsed with column specification:
cols(
pet = col_character(),
gene_A = col_double(),
gene_B = col_double(),
gene_C = col_double()
)
Parsed with column specification:
cols(
pet = col_character(),
gene_A = col_double(),
gene_B = col_double(),
gene_C = col_double()
)
| visit | pet | gene_A | gene_B | gene_C |
|---|
| 1 | David | -0.59425311 | -1.2262407 | -0.30771429 |
| 1 | Ed | -1.17645232 | -0.5087640 | 0.02835355 |
| 1 | Fiona | 0.36658576 | -0.3217073 | -1.82700451 |
| 2 | David | 0.08382534 | 0.5735718 | 0.66994801 |
| 2 | Ed | -0.40047124 | -0.1817827 | 0.92567540 |
| 2 | Fiona | 1.72076557 | 1.6649228 | 0.51074184 |
UNC protein expression
Parsed with column specification:
cols(
pet = col_character(),
prot_A = col_double(),
prot_B = col_double(),
prot_C = col_double()
)
Parsed with column specification:
cols(
pet = col_character(),
prot_A = col_double(),
prot_B = col_double(),
prot_C = col_double()
)
| visit | pet | prot_A | prot_B | prot_C |
|---|
| 1 | David | 1.04580136 | 0.1280673 | -0.6867668 |
| 1 | Ed | -1.65977553 | 0.2117860 | 0.9772767 |
| 1 | Fiona | 0.05811210 | -0.6413509 | -1.4593511 |
| 2 | David | -0.44575884 | -0.7384114 | 0.9175770 |
| 2 | Ed | 0.02765904 | 1.8317201 | 0.6595148 |
| 2 | Fiona | 0.97396187 | -0.7918110 | -0.4082508 |
Find the change in each gene from visit 1 to visit 2
| visit | pet | gene_A | gene_B | gene_C |
|---|
| 1 | Ann | -1.0073042 | -1.2805845 | -0.4479685 |
| 1 | Bob | -1.2335142 | -0.3723877 | -1.4327190 |
| 1 | Charlie | 0.6181059 | -0.8868438 | -0.4357137 |
| 2 | Ann | 0.7318518 | 0.4364545 | 0.7449587 |
| 2 | Bob | 1.2034879 | 1.1789234 | 1.4050414 |
| 2 | Charlie | -0.3126272 | 0.9244382 | 0.1664011 |
| visit | pet | gene | value |
|---|
| 1 | Ann | gene_A | -1.0073042 |
| 1 | Bob | gene_A | -1.2335142 |
| 1 | Charlie | gene_A | 0.6181059 |
| 2 | Ann | gene_A | 0.7318518 |
| 2 | Bob | gene_A | 1.2034879 |
| 2 | Charlie | gene_A | -0.3126272 |
| pet | gene | 1 | 2 |
|---|
| Ann | gene_A | -1.0073042 | 0.7318518 |
| Ann | gene_B | -1.2805845 | 0.4364545 |
| Ann | gene_C | -0.4479685 | 0.7449587 |
| Bob | gene_A | -1.2335142 | 1.2034879 |
| Bob | gene_B | -0.3723877 | 1.1789234 |
| Bob | gene_C | -1.4327190 | 1.4050414 |
| patient | gene | gene.change |
|---|
| Ann | A | 1.739156 |
| Ann | B | 1.717039 |
| Ann | C | 1.192927 |
| Bob | A | 2.437002 |
| Bob | B | 1.551311 |
| Bob | C | 2.837760 |
Combine with UNC data
| patient | gene | gene.change |
|---|
| David | A | 0.6780785 |
| David | B | 1.7998124 |
| David | C | 0.9776623 |
| Ed | A | 0.7759811 |
| Ed | B | 0.3269814 |
| Ed | C | 0.8973218 |
| site | patient | gene | gene.change |
|---|
| duke | Ann | A | 1.7391560 |
| duke | Ann | B | 1.7170389 |
| duke | Ann | C | 1.1929272 |
| duke | Bob | A | 2.4370022 |
| duke | Bob | B | 1.5513112 |
| duke | Bob | C | 2.8377604 |
| duke | Charlie | A | -0.9307330 |
| duke | Charlie | B | 1.8112820 |
| duke | Charlie | C | 0.6021148 |
| unc | David | A | 0.6780785 |
| unc | David | B | 1.7998124 |
| unc | David | C | 0.9776623 |
| unc | Ed | A | 0.7759811 |
| unc | Ed | B | 0.3269814 |
| unc | Ed | C | 0.8973218 |
| unc | Fiona | A | 1.3541798 |
| unc | Fiona | B | 1.9866301 |
| unc | Fiona | C | 2.3377464 |
Now do the same with protein data
| patient | protein | protein.change |
|---|
| Ann | A | 0.07755833 |
| Ann | B | -0.60571547 |
| Ann | C | 2.26166848 |
| Bob | A | 2.80037479 |
| Bob | B | 0.64111624 |
| Bob | C | 0.95898869 |
| patient | protein | protein.change |
|---|
| David | A | -1.4915602 |
| David | B | -0.8664787 |
| David | C | 1.6043438 |
| Ed | A | 1.6874346 |
| Ed | B | 1.6199340 |
| Ed | C | -0.3177619 |
| site | patient | protein | protein.change |
|---|
| duke | Ann | A | 0.07755833 |
| duke | Ann | B | -0.60571547 |
| duke | Ann | C | 2.26166848 |
| duke | Bob | A | 2.80037479 |
| duke | Bob | B | 0.64111624 |
| duke | Bob | C | 0.95898869 |
| duke | Charlie | A | 0.83316369 |
| duke | Charlie | B | 2.07089865 |
| duke | Charlie | C | 0.35482775 |
| unc | David | A | -1.49156020 |
| unc | David | B | -0.86647868 |
| unc | David | C | 1.60434377 |
| unc | Ed | A | 1.68743457 |
| unc | Ed | B | 1.61993404 |
| unc | Ed | C | -0.31776193 |
| unc | Fiona | A | 0.91584977 |
| unc | Fiona | B | -0.15046013 |
| unc | Fiona | C | 1.05110029 |
Combine gene and protein data into single data frame
Since the rows are perfectly aligned, we can just bind columns together.
If we are not certain that the rows match up exactly, it is safer to use
a join to combine columns.
| site | patient | gene | gene.change | site1 | patient1 | protein | protein.change |
|---|
| duke | Ann | A | 1.7391560 | duke | Ann | A | 0.07755833 |
| duke | Ann | B | 1.7170389 | duke | Ann | B | -0.60571547 |
| duke | Ann | C | 1.1929272 | duke | Ann | C | 2.26166848 |
| duke | Bob | A | 2.4370022 | duke | Bob | A | 2.80037479 |
| duke | Bob | B | 1.5513112 | duke | Bob | B | 0.64111624 |
| duke | Bob | C | 2.8377604 | duke | Bob | C | 0.95898869 |
| duke | Charlie | A | -0.9307330 | duke | Charlie | A | 0.83316369 |
| duke | Charlie | B | 1.8112820 | duke | Charlie | B | 2.07089865 |
| duke | Charlie | C | 0.6021148 | duke | Charlie | C | 0.35482775 |
| unc | David | A | 0.6780785 | unc | David | A | -1.49156020 |
| unc | David | B | 1.7998124 | unc | David | B | -0.86647868 |
| unc | David | C | 0.9776623 | unc | David | C | 1.60434377 |
| unc | Ed | A | 0.7759811 | unc | Ed | A | 1.68743457 |
| unc | Ed | B | 0.3269814 | unc | Ed | B | 1.61993404 |
| unc | Ed | C | 0.8973218 | unc | Ed | C | -0.31776193 |
| unc | Fiona | A | 1.3541798 | unc | Fiona | A | 0.91584977 |
| unc | Fiona | B | 1.9866301 | unc | Fiona | B | -0.15046013 |
| unc | Fiona | C | 2.3377464 | unc | Fiona | C | 1.05110029 |
| site | patient | gene | gene.change | protein.change |
|---|
| duke | Ann | A | 1.739156 | 0.07755833 |
| duke | Ann | B | 1.717039 | -0.60571547 |
| duke | Ann | C | 1.192927 | 2.26166848 |
| duke | Bob | A | 2.437002 | 2.80037479 |
| duke | Bob | B | 1.551311 | 0.64111624 |
| duke | Bob | C | 2.837760 | 0.95898869 |
Combine with demographic data
Parsed with column specification:
cols(
patients = col_character(),
age = col_integer(),
sex = col_character()
)
Parsed with column specification:
cols(
patients = col_character(),
age = col_integer(),
sex = col_character()
)
| patients | age | sex |
|---|
| Ann | 16 | F |
| Bob | 23 | M |
| Beth | 43 | F |
| Charlie | 53 | M |
| patients | age | sex |
|---|
| David | 49 | K |
| Edward | 53 | M |
| Fiona | 67 | F |
| site | patients | age | sex |
|---|
| duke | Ann | 16 | F |
| duke | Bob | 23 | M |
| duke | Beth | 43 | F |
| duke | Charlie | 53 | M |
| unc | David | 49 | K |
| unc | Edward | 53 | M |
| unc | Fiona | 67 | F |
| site | patient | gene | gene.change | protein.change | age | sex |
|---|
| duke | Ann | A | 1.7391560 | 0.07755833 | 16 | F |
| duke | Ann | B | 1.7170389 | -0.60571547 | 16 | F |
| duke | Ann | C | 1.1929272 | 2.26166848 | 16 | F |
| duke | Bob | A | 2.4370022 | 2.80037479 | 23 | M |
| duke | Bob | B | 1.5513112 | 0.64111624 | 23 | M |
| duke | Bob | C | 2.8377604 | 0.95898869 | 23 | M |
| duke | Charlie | A | -0.9307330 | 0.83316369 | 53 | M |
| duke | Charlie | B | 1.8112820 | 2.07089865 | 53 | M |
| duke | Charlie | C | 0.6021148 | 0.35482775 | 53 | M |
| unc | David | A | 0.6780785 | -1.49156020 | 49 | K |
| unc | David | B | 1.7998124 | -0.86647868 | 49 | K |
| unc | David | C | 0.9776623 | 1.60434377 | 49 | K |
| unc | Ed | A | 0.7759811 | 1.68743457 | NA | NA |
| unc | Ed | B | 0.3269814 | 1.61993404 | NA | NA |
| unc | Ed | C | 0.8973218 | -0.31776193 | NA | NA |
| unc | Fiona | A | 1.3541798 | 0.91584977 | 67 | F |
| unc | Fiona | B | 1.9866301 | -0.15046013 | 67 | F |
| unc | Fiona | C | 2.3377464 | 1.05110029 | 67 | F |
There are missing values for Ed because the names don’t match. We know
that Ed’s name in this case is really Edward, so we fix and try again.
| site | patient | gene | gene.change | protein.change | age | sex |
|---|
| duke | Ann | A | 1.7391560 | 0.07755833 | 16 | F |
| duke | Ann | B | 1.7170389 | -0.60571547 | 16 | F |
| duke | Ann | C | 1.1929272 | 2.26166848 | 16 | F |
| duke | Bob | A | 2.4370022 | 2.80037479 | 23 | M |
| duke | Bob | B | 1.5513112 | 0.64111624 | 23 | M |
| duke | Bob | C | 2.8377604 | 0.95898869 | 23 | M |
| duke | Charlie | A | -0.9307330 | 0.83316369 | 53 | M |
| duke | Charlie | B | 1.8112820 | 2.07089865 | 53 | M |
| duke | Charlie | C | 0.6021148 | 0.35482775 | 53 | M |
| unc | David | A | 0.6780785 | -1.49156020 | 49 | K |
| unc | David | B | 1.7998124 | -0.86647868 | 49 | K |
| unc | David | C | 0.9776623 | 1.60434377 | 49 | K |
| unc | Edward | A | 0.7759811 | 1.68743457 | 53 | M |
| unc | Edward | B | 0.3269814 | 1.61993404 | 53 | M |
| unc | Edward | C | 0.8973218 | -0.31776193 | 53 | M |
| unc | Fiona | A | 1.3541798 | 0.91584977 | 67 | F |
| unc | Fiona | B | 1.9866301 | -0.15046013 | 67 | F |
| unc | Fiona | C | 2.3377464 | 1.05110029 | 67 | F |
| site | patient | gene | gene.change | protein.change | age | sex |
|---|
| duke | Ann | A | 1.739156 | 0.07755833 | 16 | F |
| duke | Ann | B | 1.717039 | -0.60571547 | 16 | F |
| duke | Ann | C | 1.192927 | 2.26166848 | 16 | F |
| duke | Bob | A | 2.437002 | 2.80037479 | 23 | M |
| duke | Bob | B | 1.551311 | 0.64111624 | 23 | M |
| duke | Bob | C | 2.837760 | 0.95898869 | 23 | M |
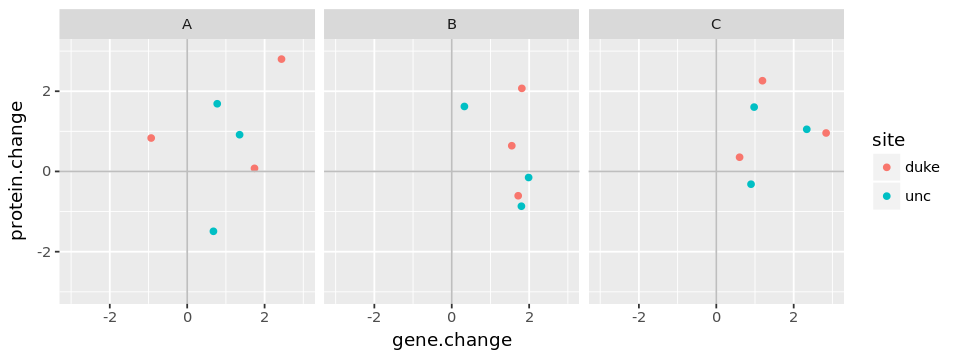
Plotting the data suggests that there may be an increase in most genes
and proteins from visit 1 to visit 2 (since most of the changes are
positive), but there is no obvious correlation between the gene and its
protein product.
Data type cannot be displayed: