Set up environment¶
In [1]:
source("pilot_config.R")
Loading tidyverse: ggplot2
Loading tidyverse: tibble
Loading tidyverse: tidyr
Loading tidyverse: readr
Loading tidyverse: purrr
Loading tidyverse: dplyr
Conflicts with tidy packages ---------------------------------------------------
filter(): dplyr, stats
lag(): dplyr, stats
Attaching package: ‘foreach’
The following objects are masked from ‘package:purrr’:
accumulate, when
Loading required package: S4Vectors
Loading required package: stats4
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:parallel’:
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from ‘package:dplyr’:
combine, intersect, setdiff, union
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, append, as.data.frame, cbind, colMeans, colnames,
colSums, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, lengths, Map, mapply, match,
mget, order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rowMeans, rownames, rowSums, sapply, setdiff, sort,
table, tapply, union, unique, unsplit, which, which.max, which.min
Attaching package: ‘S4Vectors’
The following objects are masked from ‘package:dplyr’:
first, rename
The following object is masked from ‘package:tidyr’:
expand
The following object is masked from ‘package:base’:
expand.grid
Loading required package: IRanges
Attaching package: ‘IRanges’
The following objects are masked from ‘package:dplyr’:
collapse, desc, slice
The following object is masked from ‘package:purrr’:
reduce
Loading required package: GenomicRanges
Loading required package: GenomeInfoDb
Loading required package: SummarizedExperiment
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: DelayedArray
Loading required package: matrixStats
Attaching package: ‘matrixStats’
The following objects are masked from ‘package:Biobase’:
anyMissing, rowMedians
The following object is masked from ‘package:dplyr’:
count
Attaching package: ‘DelayedArray’
The following objects are masked from ‘package:matrixStats’:
colMaxs, colMins, colRanges, rowMaxs, rowMins, rowRanges
The following object is masked from ‘package:base’:
apply
Attaching package: ‘limma’
The following object is masked from ‘package:DESeq2’:
plotMA
The following object is masked from ‘package:BiocGenerics’:
plotMA
Attaching package: ‘gridExtra’
The following object is masked from ‘package:Biobase’:
combine
The following object is masked from ‘package:BiocGenerics’:
combine
The following object is masked from ‘package:dplyr’:
combine
---------------------
Welcome to dendextend version 1.8.0
Type citation('dendextend') for how to cite the package.
Type browseVignettes(package = 'dendextend') for the package vignette.
The github page is: https://github.com/talgalili/dendextend/
Suggestions and bug-reports can be submitted at: https://github.com/talgalili/dendextend/issues
Or contact: <tal.galili@gmail.com>
To suppress this message use: suppressPackageStartupMessages(library(dendextend))
---------------------
Attaching package: ‘dendextend’
The following object is masked from ‘package:stats’:
cutree
Attaching package: ‘plotly’
The following object is masked from ‘package:IRanges’:
slice
The following object is masked from ‘package:S4Vectors’:
rename
The following object is masked from ‘package:ggplot2’:
last_plot
The following object is masked from ‘package:stats’:
filter
The following object is masked from ‘package:graphics’:
layout
Recall that after the tutorial one, we have created the hts-pilot-2018.RData.
scratch
└── analysis_output
├── out
│ └── hts-pilot-2018.RData
└── img
In [2]:
OUTDIR
'/home/jovyan/work/scratch/analysis_output/out'
In [3]:
# file directory
cntfile <- file.path(OUTDIR, "hts-pilot-2018.RData")
Read in results¶
In [4]:
### Import count data
attach(cntfile)
tools::md5sum(cntfile)
/home/jovyan/work/scratch/analysis_output/out/hts-pilot-2018.RData: 'e8d7991f674b7a9284abae8f38656c2e'
In [5]:
### Import metadata
tools::md5sum(METADTFILE)
mtdf <- readr::read_tsv(METADTFILE) %>%
mutate_at(vars(
`Label`,
`Strain`,
`Media`,
`enrichment_method`,
`library#`,
`libprep_person`,
`experiment_person`
), factor)
/home/jovyan/work/HTS2018-notebooks/josh/info/2018_pilot_metadata_anon.tsv: '20b31fdc4487d0cd8cc3c9300584faca'
Parsed with column specification:
cols(
Label = col_character(),
RNA_sample_num = col_integer(),
Media = col_character(),
Strain = col_character(),
Replicate = col_integer(),
experiment_person = col_character(),
libprep_person = col_character(),
enrichment_method = col_character(),
RIN = col_double(),
concentration_fold_difference = col_double(),
`i7 index` = col_character(),
`i5 index` = col_character(),
`i5 primer` = col_character(),
`i7 primer` = col_character(),
`library#` = col_integer()
)
Recall that there are 204 samples and 8498 genes
In [6]:
head(mtdf)
| Label | RNA_sample_num | Media | Strain | Replicate | experiment_person | libprep_person | enrichment_method | RIN | concentration_fold_difference | i7 index | i5 index | i5 primer | i7 primer | library# |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2_MA_C | 2 | YPD | H99 | 2 | expA | prepA | MA | 10.0 | 1.34 | ATTACTCG | AGGCTATA | i501 | i701 | 1 |
| 9_MA_C | 9 | YPD | mar1d | 3 | expA | prepA | MA | 10.0 | 2.23 | ATTACTCG | GCCTCTAT | i502 | i701 | 2 |
| 10_MA_C | 10 | YPD | mar1d | 4 | expA | prepA | MA | 9.9 | 4.37 | ATTACTCG | AGGATAGG | i503 | i701 | 3 |
| 14_MA_C | 14 | TC | H99 | 2 | expA | prepA | MA | 10.0 | 1.57 | ATTACTCG | TCAGAGCC | i504 | i701 | 4 |
| 15_MA_C | 15 | TC | H99 | 3 | expA | prepA | MA | 9.9 | 2.85 | ATTACTCG | CTTCGCCT | i505 | i701 | 5 |
| 21_MA_C | 21 | TC | mar1d | 3 | expA | prepA | MA | 10.0 | 1.81 | ATTACTCG | TAAGATTA | i506 | i701 | 6 |
Check the label between metadata and mapping results¶
Recall that we got two data frames in the previous tutorial:
- genecounts: gene counts for each sample - Note: We will need to
convert it into gene counts for each library later - mapresults:
the mapping results - Note: There are 204 samples
The metadata (mtdf) contains the information of each sample. Here we
need to make sure if the label in the metadata matches the sample names
we have in mapresults and genecount
The code chunk below allows us to check to see if we can match the labels to the those in the metadata file. There should not be any output from the code chunk.
In [7]:
### Use setdiff to check to see if we can match the labels to the those in the metadata file
myregex <- "_[A-Z](100|[1-9][0-9]?)_L00[1-4].*"
mapresults$expid %>%
str_replace(myregex, "") %>%
setdiff(mtdf$Label)
mtdf$Label %>%
setdiff(str_replace(mapresults$expid, myregex, ""))
Construct gene count matrix for each library¶
Add the “Label” to the count matrix and mapping results, then merge in phenotype data (by Label)
In [8]:
### Add "Label" to genecounts
genecounts %>%
mutate(Label = str_replace(expid, myregex, "")) ->
annogenecnts
In [9]:
### Collapse the gene counts within each label
annogenecnts %>%
select(-expid) %>%
group_by(Label) %>%
summarize_all(sum) %>%
gather(gene, value, -Label) %>%
spread(Label, value) ->
annogenecnts0
Show the resulting data frame in each step
In [10]:
genecounts[1:6, 1:6]
| expid | CNAG_00001 | CNAG_00002 | CNAG_00003 | CNAG_00004 | CNAG_00005 |
|---|---|---|---|---|---|
| 1_MA_J_S18_L001_ReadsPerGene.out.tab | 0 | 66 | 38 | 74 | 33 |
| 1_MA_J_S18_L002_ReadsPerGene.out.tab | 0 | 59 | 25 | 79 | 25 |
| 1_MA_J_S18_L003_ReadsPerGene.out.tab | 0 | 74 | 27 | 79 | 32 |
| 1_MA_J_S18_L004_ReadsPerGene.out.tab | 0 | 66 | 22 | 69 | 24 |
| 1_RZ_J_S26_L001_ReadsPerGene.out.tab | 0 | 50 | 16 | 51 | 26 |
| 1_RZ_J_S26_L002_ReadsPerGene.out.tab | 0 | 45 | 7 | 51 | 31 |
In [11]:
annogenecnts[1:6, c(colnames(annogenecnts)[1:6], "Label")]
| expid | CNAG_00001 | CNAG_00002 | CNAG_00003 | CNAG_00004 | CNAG_00005 | Label |
|---|---|---|---|---|---|---|
| 1_MA_J_S18_L001_ReadsPerGene.out.tab | 0 | 66 | 38 | 74 | 33 | 1_MA_J |
| 1_MA_J_S18_L002_ReadsPerGene.out.tab | 0 | 59 | 25 | 79 | 25 | 1_MA_J |
| 1_MA_J_S18_L003_ReadsPerGene.out.tab | 0 | 74 | 27 | 79 | 32 | 1_MA_J |
| 1_MA_J_S18_L004_ReadsPerGene.out.tab | 0 | 66 | 22 | 69 | 24 | 1_MA_J |
| 1_RZ_J_S26_L001_ReadsPerGene.out.tab | 0 | 50 | 16 | 51 | 26 | 1_RZ_J |
| 1_RZ_J_S26_L002_ReadsPerGene.out.tab | 0 | 45 | 7 | 51 | 31 | 1_RZ_J |
In [12]:
annogenecnts0[1:6, 1:6]
| gene | 1_MA_J | 1_RZ_J | 10_MA_C | 10_RZ_C | 11_MA_J |
|---|---|---|---|---|---|
| CNAG_00001 | 0 | 0 | 0 | 0 | 0 |
| CNAG_00002 | 265 | 204 | 269 | 76 | 130 |
| CNAG_00003 | 112 | 40 | 171 | 24 | 124 |
| CNAG_00004 | 301 | 207 | 407 | 141 | 272 |
| CNAG_00005 | 114 | 125 | 50 | 25 | 38 |
| CNAG_00006 | 1904 | 1295 | 3571 | 1015 | 2073 |
Metadata¶
Add “Label” to read map results and merge in phenotype data (-> annomapres)
In [13]:
mapresults %>%
mutate(Label = str_replace(expid, myregex, "")) %>%
full_join(mtdf, by = "Label") ->
annomapres
Warning message:
“Column `Label` joining character vector and factor, coercing into character vector”
In [14]:
grpvars <- vars(Label, Strain, Media, experiment_person, libprep_person, enrichment_method)
sumvars <- vars(prop.gene, prop.nofeat, prop.unique, depth)
annomapres %>%
group_by_at(grpvars) %>%
summarize_at(sumvars, mean) ->
annomapres0
In [15]:
head(annomapres)
| expid | ngenemap | namb | nmulti | nnofeat | nunmap | depth | prop.gene | prop.nofeat | prop.unique | ⋯ | experiment_person | libprep_person | enrichment_method | RIN | concentration_fold_difference | i7 index | i5 index | i5 primer | i7 primer | library# |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1_MA_J_S18_L001_ReadsPerGene.out.tab | 2399781 | 647 | 66100 | 20347 | 2690 | 2489565 | 0.9639359 | 0.008172914 | 0.9721088 | ⋯ | expA | prepB | MA | 10 | 3.64 | CGCTCATT | AGGCTATA | i501 | i703 | 18 |
| 1_MA_J_S18_L002_ReadsPerGene.out.tab | 2362228 | 652 | 65234 | 20004 | 2684 | 2450802 | 0.9638592 | 0.008162226 | 0.9720214 | ⋯ | expA | prepB | MA | 10 | 3.64 | CGCTCATT | AGGCTATA | i501 | i703 | 18 |
| 1_MA_J_S18_L003_ReadsPerGene.out.tab | 2436776 | 697 | 66538 | 20549 | 2672 | 2527232 | 0.9642075 | 0.008131030 | 0.9723385 | ⋯ | expA | prepB | MA | 10 | 3.64 | CGCTCATT | AGGCTATA | i501 | i703 | 18 |
| 1_MA_J_S18_L004_ReadsPerGene.out.tab | 2417485 | 616 | 65066 | 20505 | 2585 | 2506257 | 0.9645798 | 0.008181523 | 0.9727614 | ⋯ | expA | prepB | MA | 10 | 3.64 | CGCTCATT | AGGCTATA | i501 | i703 | 18 |
| 1_RZ_J_S26_L001_ReadsPerGene.out.tab | 2366742 | 1431 | 395848 | 768146 | 7218 | 3539385 | 0.6686874 | 0.217028100 | 0.8857155 | ⋯ | expA | prepB | RZ | 10 | 3.64 | GAGATTCC | AGGCTATA | i501 | i704 | 26 |
| 1_RZ_J_S26_L002_ReadsPerGene.out.tab | 2331658 | 1337 | 388079 | 755654 | 7022 | 3483750 | 0.6692954 | 0.216908217 | 0.8862037 | ⋯ | expA | prepB | RZ | 10 | 3.64 | GAGATTCC | AGGCTATA | i501 | i704 | 26 |
In [16]:
head(annomapres0)
| Label | Strain | Media | experiment_person | libprep_person | enrichment_method | prop.gene | prop.nofeat | prop.unique | depth |
|---|---|---|---|---|---|---|---|---|---|
| 1_MA_J | H99 | YPD | expA | prepB | MA | 0.9641456 | 0.008161923 | 0.9723075 | 2493464 |
| 1_RZ_J | H99 | YPD | expA | prepB | RZ | 0.6689001 | 0.217095621 | 0.8859957 | 3541358 |
| 10_MA_C | mar1d | YPD | expA | prepA | MA | 0.9618651 | 0.009818573 | 0.9716837 | 3282785 |
| 10_RZ_C | mar1d | YPD | expA | prepA | RZ | 0.7497438 | 0.200651686 | 0.9503955 | 1742594 |
| 11_MA_J | mar1d | YPD | expA | prepB | MA | 0.9669597 | 0.008717898 | 0.9756776 | 2062181 |
| 11_RZ_J | mar1d | YPD | expA | prepB | RZ | 0.7030020 | 0.195547151 | 0.8985491 | 2621913 |
Store the results¶
In [17]:
outfile <- file.path(OUTDIR, "HTS-Pilot-Annotated-STAR-counts.RData")
save(mtdf, annogenecnts0, annomapres0, annogenecnts, annomapres, file = outfile)
tools::md5sum(outfile)
/home/jovyan/work/scratch/analysis_output/out/HTS-Pilot-Annotated-STAR-counts.RData: '79338a9ad2efa4bcd1e6e9fd2a2b0ef7'
Visualize the mapping results¶
In [18]:
### Figures for mapping results
mygeom <- geom_point(position = position_jitter(w = 0.3, h = 0))
mypal <- scale_colour_manual(name="", values =brewer.pal(3,"Set1"))
mytheme <- theme(axis.text.x = element_text(angle = 90, hjust = 1)) + theme_bw()
myfacet <- facet_grid(Strain~ Media, drop=TRUE, scales="free_x", space="free")
Show the fraction of unique mapped reads among all reads (prop.unique)¶
In [19]:
options(repr.plot.width = 9, repr.plot.height = 6)
p1 <- ggplot(annomapres,
aes(x = factor(Label),
y = prop.unique,
shape = Strain,
color = Media)) +
myfacet +
mygeom +
mytheme +
mypal
print(p1)
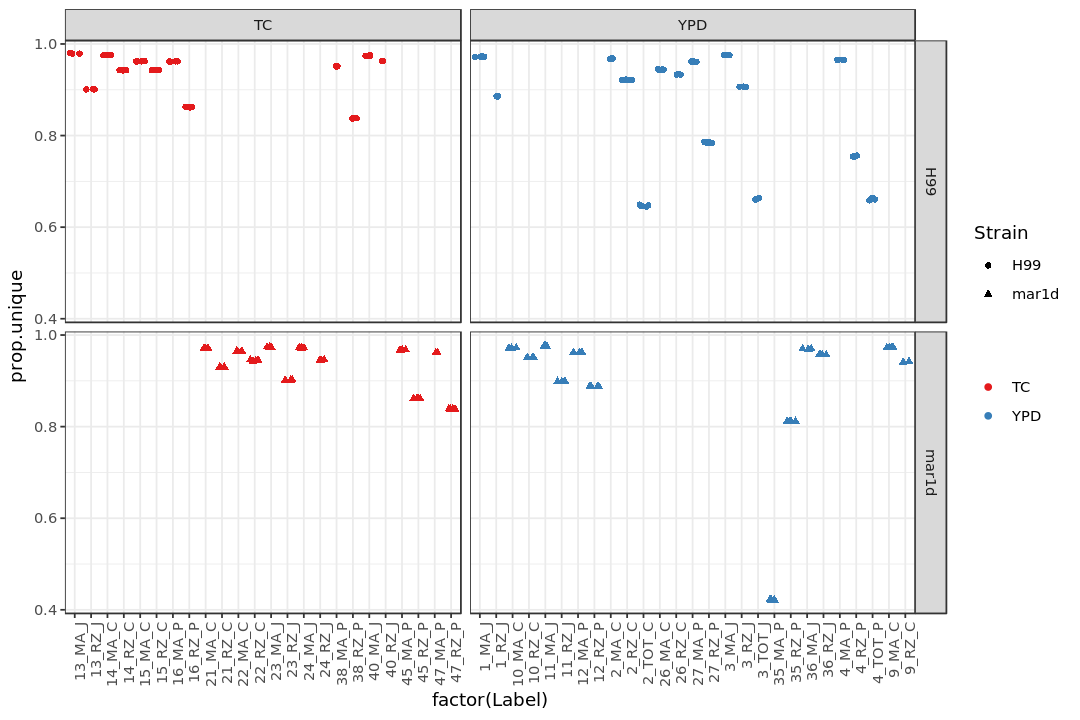
Show the fraction of reads mapped to genes (prop.gene)¶
In [20]:
options(repr.plot.width = 9, repr.plot.height = 6)
p2 <- ggplot(annomapres,
aes(x = Label,
y = prop.gene,
shape = Strain,
color = Media)) +
myfacet +
mygeom +
mytheme +
mypal
print(p2)
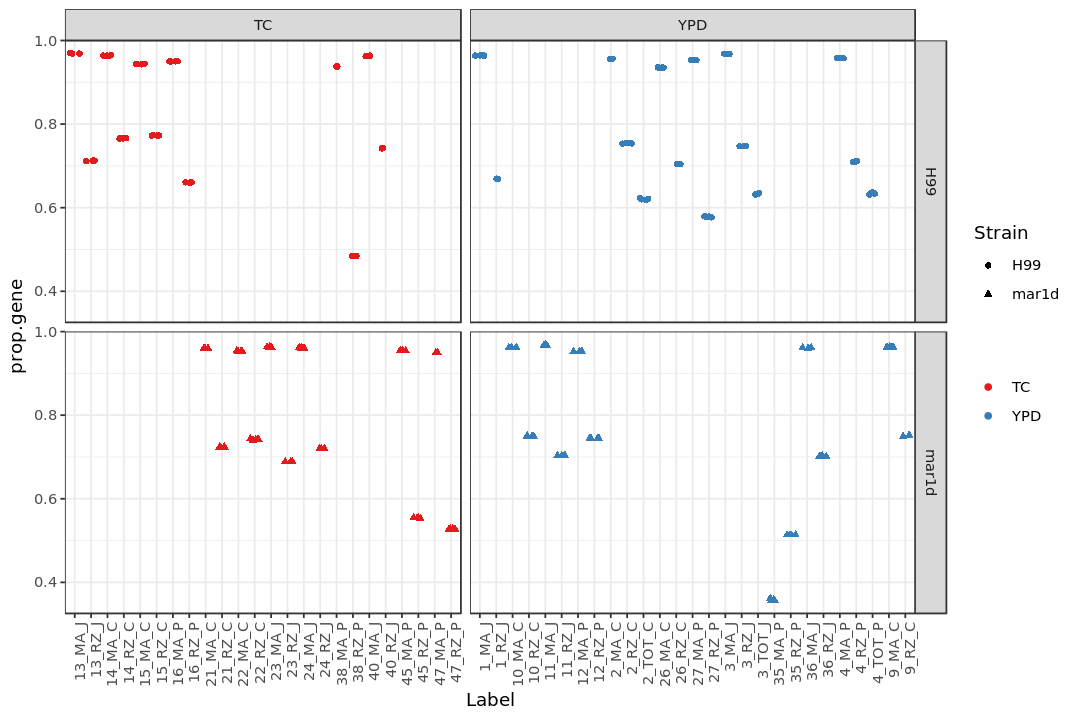
Show the fraction of reads categorized as “no feature” (prop.nofeat)¶
In [21]:
options(repr.plot.width = 9, repr.plot.height = 6)
p3 <- ggplot(annomapres,
aes(x = Label,
y = prop.nofeat,
shape = Strain,
color = Media))+
myfacet +
mygeom +
mytheme +
mypal
print(p3)
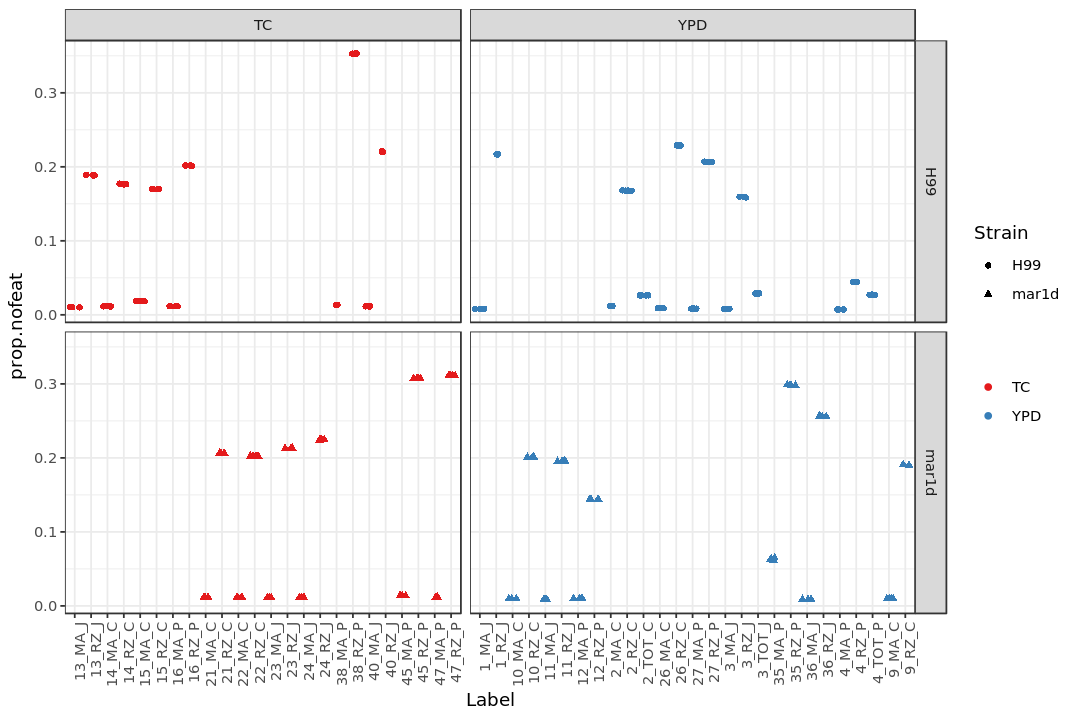
Show the number of all the reads in each sample¶
(Note: depth = ngenemap + namb + nmulti + nnofeat + nunmap)
In [22]:
options(repr.plot.width = 9, repr.plot.height = 6)
p4 <- ggplot(annomapres,
aes(x = Label,
y = depth,
shape = Strain,
color = Media))+
myfacet +
mygeom +
mytheme +
mypal
print(p4)
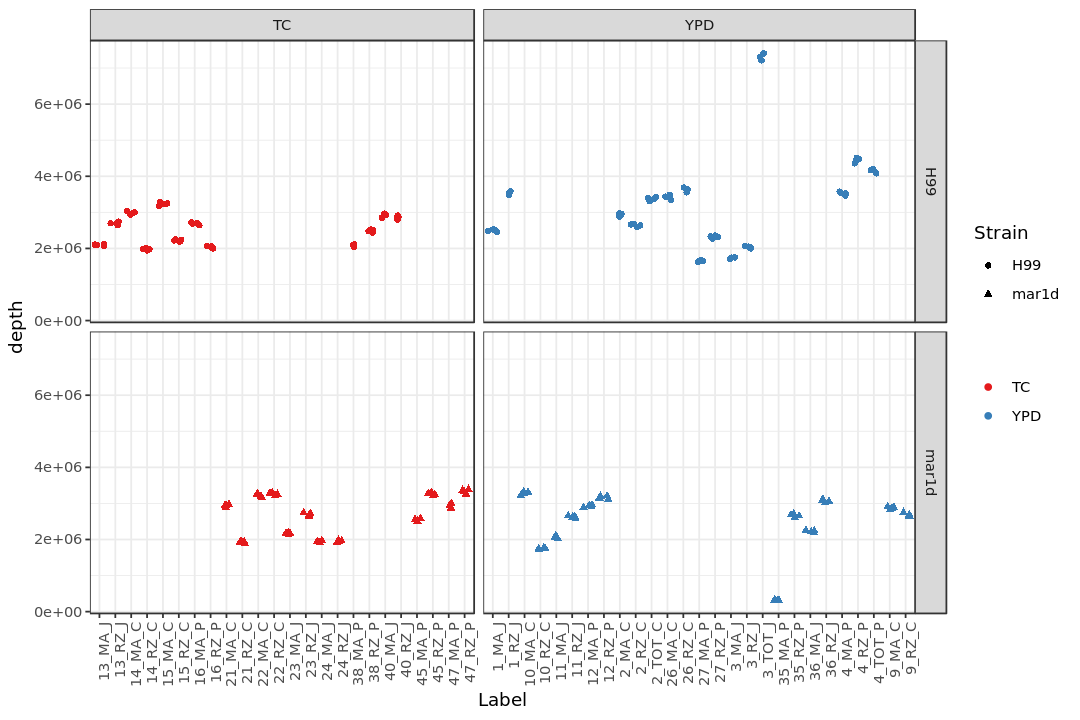
Store the plots¶
In [23]:
png(file.path(IMGDIR, "p1.png"), height = 480 * 2, width = 480 * 2)
plot(p1)
graphics.off()
png(file.path(IMGDIR, "p2.png"), height = 480 * 2, width = 480 * 2)
plot(p2)
graphics.off()
png(file.path(IMGDIR, "p3.png"), height = 480 * 2, width = 480 * 2)
plot(p3)
graphics.off()
png(file.path(IMGDIR, "p4.png"), height = 480 * 2, width = 480 * 2)
plot(p4)
graphics.off()
The End¶
In [24]:
sessionInfo()
R version 3.4.1 (2017-06-30)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Debian GNU/Linux 8 (jessie)
Matrix products: default
BLAS: /opt/conda/lib/R/lib/libRblas.so
LAPACK: /opt/conda/lib/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] tools parallel stats4 stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] bindrcpp_0.2 plotly_4.8.0
[3] dendextend_1.8.0 gridExtra_2.3
[5] RColorBrewer_1.1-2 qvalue_2.10.0
[7] limma_3.34.9 DESeq2_1.18.1
[9] SummarizedExperiment_1.8.0 DelayedArray_0.4.1
[11] matrixStats_0.53.1 Biobase_2.38.0
[13] GenomicRanges_1.30.3 GenomeInfoDb_1.14.0
[15] IRanges_2.12.0 S4Vectors_0.16.0
[17] BiocGenerics_0.24.0 haven_1.1.1
[19] stringr_1.3.0 foreach_1.4.4
[21] dplyr_0.7.4 purrr_0.2.4
[23] readr_1.1.1 tidyr_0.8.1
[25] tibble_1.4.2 ggplot2_3.0.0
[27] tidyverse_1.1.1
loaded via a namespace (and not attached):
[1] colorspace_1.3-2 class_7.3-14 modeltools_0.2-22
[4] mclust_5.4.1 IRdisplay_0.4.4 htmlTable_1.9
[7] XVector_0.18.0 base64enc_0.1-3 flexmix_2.3-14
[10] bit64_0.9-5 mvtnorm_1.0-8 AnnotationDbi_1.40.0
[13] lubridate_1.7.4 xml2_1.2.0 codetools_0.2-15
[16] splines_3.4.1 mnormt_1.5-5 robustbase_0.92-7
[19] geneplotter_1.56.0 knitr_1.20 IRkernel_0.8.11
[22] Formula_1.2-1 jsonlite_1.5 broom_0.4.4
[25] annotate_1.56.0 kernlab_0.9-25 cluster_2.0.6
[28] compiler_3.4.1 httr_1.3.1 backports_1.1.2
[31] assertthat_0.2.0 Matrix_1.2-14 lazyeval_0.2.1
[34] acepack_1.4.1 htmltools_0.3.6 gtable_0.2.0
[37] glue_1.2.0 GenomeInfoDbData_0.99.0 reshape2_1.4.3
[40] Rcpp_0.12.15 trimcluster_0.1-2.1 cellranger_1.1.0
[43] nlme_3.1-131 fpc_2.1-11.1 iterators_1.0.9
[46] psych_1.8.4 rvest_0.3.2 XML_3.98-1.11
[49] DEoptimR_1.0-8 MASS_7.3-48 zlibbioc_1.24.0
[52] scales_0.5.0 hms_0.3 memoise_1.1.0
[55] rpart_4.1-13 latticeExtra_0.6-28 stringi_1.1.7
[58] RSQLite_2.0 genefilter_1.60.0 checkmate_1.8.5
[61] BiocParallel_1.12.0 repr_0.15.0 prabclus_2.2-6
[64] rlang_0.2.1 pkgconfig_2.0.1 bitops_1.0-6
[67] evaluate_0.10.1 lattice_0.20-34 bindr_0.1.1
[70] labeling_0.3 htmlwidgets_1.2 tidyselect_0.2.4
[73] bit_1.1-12 plyr_1.8.4 magrittr_1.5
[76] R6_2.2.2 Hmisc_4.0-3 pbdZMQ_0.3-2
[79] DBI_1.0.0 pillar_1.2.2 whisker_0.3-2
[82] foreign_0.8-67 withr_2.1.1 survival_2.40-1
[85] RCurl_1.95-4.8 nnet_7.3-12 modelr_0.1.2
[88] crayon_1.3.4 uuid_0.1-2 viridis_0.5.1
[91] locfit_1.5-9.1 grid_3.4.1 readxl_1.1.0
[94] data.table_1.10.4 blob_1.1.1 forcats_0.3.0
[97] diptest_0.75-7 digest_0.6.15 xtable_1.8-2
[100] munsell_0.5.0 viridisLite_0.3.0