Set environment¶
In [1]:
# load required libraries
library(DESeq2)
library(tidyverse)
library(RColorBrewer)
library(dendextend)
Loading required package: S4Vectors
Loading required package: stats4
Loading required package: BiocGenerics
Loading required package: parallel
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:parallel’:
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLB
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, append, as.data.frame, cbind, colMeans, colnames,
colSums, do.call, duplicated, eval, evalq, Filter, Find, get, grep,
grepl, intersect, is.unsorted, lapply, lengths, Map, mapply, match,
mget, order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
rbind, Reduce, rowMeans, rownames, rowSums, sapply, setdiff, sort,
table, tapply, union, unique, unsplit, which, which.max, which.min
Attaching package: ‘S4Vectors’
The following object is masked from ‘package:base’:
expand.grid
Loading required package: IRanges
Loading required package: GenomicRanges
Loading required package: GenomeInfoDb
Loading required package: SummarizedExperiment
Loading required package: Biobase
Welcome to Bioconductor
Vignettes contain introductory material; view with
'browseVignettes()'. To cite Bioconductor, see
'citation("Biobase")', and for packages 'citation("pkgname")'.
Loading required package: DelayedArray
Loading required package: matrixStats
Attaching package: ‘matrixStats’
The following objects are masked from ‘package:Biobase’:
anyMissing, rowMedians
Attaching package: ‘DelayedArray’
The following objects are masked from ‘package:matrixStats’:
colMaxs, colMins, colRanges, rowMaxs, rowMins, rowRanges
The following object is masked from ‘package:base’:
apply
Loading tidyverse: ggplot2
Loading tidyverse: tibble
Loading tidyverse: tidyr
Loading tidyverse: readr
Loading tidyverse: purrr
Loading tidyverse: dplyr
Conflicts with tidy packages ---------------------------------------------------
collapse(): dplyr, IRanges
combine(): dplyr, Biobase, BiocGenerics
count(): dplyr, matrixStats
desc(): dplyr, IRanges
expand(): tidyr, S4Vectors
filter(): dplyr, stats
first(): dplyr, S4Vectors
lag(): dplyr, stats
Position(): ggplot2, BiocGenerics, base
reduce(): purrr, GenomicRanges, IRanges
rename(): dplyr, S4Vectors
slice(): dplyr, IRanges
---------------------
Welcome to dendextend version 1.8.0
Type citation('dendextend') for how to cite the package.
Type browseVignettes(package = 'dendextend') for the package vignette.
The github page is: https://github.com/talgalili/dendextend/
Suggestions and bug-reports can be submitted at: https://github.com/talgalili/dendextend/issues
Or contact: <tal.galili@gmail.com>
To suppress this message use: suppressPackageStartupMessages(library(dendextend))
---------------------
Attaching package: ‘dendextend’
The following object is masked from ‘package:stats’:
cutree
Your Folder Structure:
└── HTS2018
├── out
│ └── hts-pilot-2018.RData
| └── HTS-Pilot-Annotated-STAR-counts.RData
└── img
In [3]:
# set directories
outdir <- "/home/jovyan/work/HTS2018/out"
imgdir <- "/home/jovyan/work/HTS2018/img"
Read in data¶
In [4]:
attach(file.path(outdir, "HTS-Pilot-Annotated-STAR-counts.RData"))
Annotated mapping results¶
In [5]:
head(annomapres0)
| Label | Strain | Media | experiment_person | libprep_person | enrichment_method | prob.gene | prob.nofeat | prob.unique | depth |
|---|---|---|---|---|---|---|---|---|---|
| 1_MA_J | H99 | YPD | S | J | MA | 0.9641456 | 0.008161923 | 0.9723075 | 2493464 |
| 1_RZ_J | H99 | YPD | S | J | RZ | 0.6689001 | 0.217095621 | 0.8859957 | 3541358 |
| 10_MA_C | mar1d | YPD | S | C | MA | 0.9618651 | 0.009818573 | 0.9716837 | 3282785 |
| 10_RZ_C | mar1d | YPD | S | C | RZ | 0.7497438 | 0.200651686 | 0.9503955 | 1742594 |
| 11_MA_J | mar1d | YPD | S | J | MA | 0.9669597 | 0.008717898 | 0.9756776 | 2062181 |
| 11_RZ_J | mar1d | YPD | S | J | RZ | 0.7030020 | 0.195547151 | 0.8985491 | 2621913 |
Annotated count matrix¶
In [6]:
head(annogenecnts0)
| gene | 1_MA_J | 1_RZ_J | 10_MA_C | 10_RZ_C | 11_MA_J | 11_RZ_J | 12_MA_P | 12_RZ_P | 13_MA_J | ⋯ | 4_RZ_P | 4_TOT_P | 40_MA_J | 40_RZ_J | 45_MA_P | 45_RZ_P | 47_MA_P | 47_RZ_P | 9_MA_C | 9_RZ_C |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CNAG_00001 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ⋯ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CNAG_00002 | 265 | 204 | 269 | 76 | 130 | 92 | 205 | 64 | 308 | ⋯ | 51 | 13 | 519 | 235 | 410 | 122 | 534 | 112 | 217 | 106 |
| CNAG_00003 | 112 | 40 | 171 | 24 | 124 | 18 | 150 | 34 | 221 | ⋯ | 11 | 8 | 218 | 53 | 232 | 46 | 240 | 41 | 128 | 35 |
| CNAG_00004 | 301 | 207 | 407 | 141 | 272 | 179 | 351 | 156 | 533 | ⋯ | 50 | 36 | 719 | 442 | 518 | 238 | 622 | 277 | 310 | 234 |
| CNAG_00005 | 114 | 125 | 50 | 25 | 38 | 32 | 38 | 12 | 202 | ⋯ | 25 | 14 | 344 | 270 | 202 | 81 | 256 | 118 | 45 | 40 |
| CNAG_00006 | 1904 | 1295 | 3571 | 1015 | 2073 | 1327 | 3003 | 926 | 1660 | ⋯ | 319 | 110 | 3002 | 1535 | 3934 | 1090 | 4966 | 1132 | 3313 | 1509 |
DESeq object¶
Generate count matrix for RZ enrichment method
In [7]:
### Prepare columnData (DataFrameand countData (matrix object)
### Select samples from RZ
annomapres0 %>%
dplyr::filter(enrichment_method == "RZ") %>%
DataFrame ->
columnData
rownames(columnData) <- columnData[["Label"]]
annogenecnts0 %>%
dplyr::select(dput(as.character(c("gene",columnData[["Label"]])))) %>%
as.data.frame %>%
column_to_rownames("gene") %>%
as.matrix ->
countData
c("gene", "1_RZ_J", "10_RZ_C", "11_RZ_J", "12_RZ_P", "13_RZ_J",
"14_RZ_C", "15_RZ_C", "16_RZ_P", "2_RZ_C", "21_RZ_C", "22_RZ_C",
"23_RZ_J", "24_RZ_J", "26_RZ_C", "27_RZ_P", "3_RZ_J", "35_RZ_P",
"36_RZ_J", "38_RZ_P", "4_RZ_P", "40_RZ_J", "45_RZ_P", "47_RZ_P",
"9_RZ_C")
In [8]:
head(countData)
| 1_RZ_J | 10_RZ_C | 11_RZ_J | 12_RZ_P | 13_RZ_J | 14_RZ_C | 15_RZ_C | 16_RZ_P | 2_RZ_C | 21_RZ_C | ⋯ | 27_RZ_P | 3_RZ_J | 35_RZ_P | 36_RZ_J | 38_RZ_P | 4_RZ_P | 40_RZ_J | 45_RZ_P | 47_RZ_P | 9_RZ_C | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CNAG_00001 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | ⋯ | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CNAG_00002 | 204 | 76 | 92 | 64 | 230 | 182 | 200 | 129 | 168 | 124 | ⋯ | 43 | 107 | 150 | 109 | 95 | 51 | 235 | 122 | 112 | 106 |
| CNAG_00003 | 40 | 24 | 18 | 34 | 56 | 53 | 54 | 40 | 40 | 41 | ⋯ | 9 | 24 | 26 | 43 | 43 | 11 | 53 | 46 | 41 | 35 |
| CNAG_00004 | 207 | 141 | 179 | 156 | 396 | 363 | 416 | 254 | 184 | 312 | ⋯ | 56 | 140 | 224 | 202 | 166 | 50 | 442 | 238 | 277 | 234 |
| CNAG_00005 | 125 | 25 | 32 | 12 | 201 | 179 | 187 | 124 | 121 | 122 | ⋯ | 22 | 67 | 154 | 56 | 82 | 25 | 270 | 81 | 118 | 40 |
| CNAG_00006 | 1295 | 1015 | 1327 | 926 | 1143 | 879 | 1058 | 619 | 1060 | 1103 | ⋯ | 398 | 827 | 869 | 1877 | 627 | 319 | 1535 | 1090 | 1132 | 1509 |
Import count matrix as a DESeq object
In [9]:
### Make DESeq object on the basis of the counts
dds <- DESeqDataSetFromMatrix(countData, columnData, ~ Media + Strain + Media:Strain)
### Estimate Size Factors
dds <- estimateSizeFactors(dds)
### Estimate Dispersion parameters (for each gene)
dds <- estimateDispersions(dds)
### Fit NB MLE model
dds <- DESeq(dds)
### Rlog "normalized" expressions
rld <- rlog(dds)
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
using pre-existing size factors
estimating dispersions
found already estimated dispersions, replacing these
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
fitting model and testing
In [10]:
### Show top 10 hits
results(dds, tidy = TRUE) %>% arrange(pvalue) %>% head(10)
| row | baseMean | log2FoldChange | lfcSE | stat | pvalue | padj |
|---|---|---|---|---|---|---|
| CNAG_04307 | 490.86196 | 4.023118 | 0.29759976 | 13.518553 | 1.215429e-41 | 9.552054e-38 |
| CNAG_05459 | 636.02477 | 1.144042 | 0.09994094 | 11.447183 | 2.429119e-30 | 9.545224e-27 |
| CNAG_03621 | 1821.27006 | -1.064434 | 0.10659585 | -9.985699 | 1.760551e-23 | 4.612056e-20 |
| CNAG_00600 | 848.44047 | 1.142434 | 0.11598632 | 9.849729 | 6.872879e-23 | 1.350349e-19 |
| CNAG_04585 | 812.31911 | -4.040747 | 0.41261597 | -9.792997 | 1.206656e-22 | 1.896623e-19 |
| CNAG_03735 | 521.96604 | 1.634231 | 0.16755798 | 9.753228 | 1.786944e-22 | 2.340599e-19 |
| CNAG_07862 | 85.91321 | -2.608844 | 0.26841617 | -9.719401 | 2.492446e-22 | 2.798304e-19 |
| CNAG_02733 | 15.32034 | -4.023640 | 0.41731949 | -9.641629 | 5.333480e-22 | 5.239478e-19 |
| CNAG_06517 | 850.38128 | 1.871991 | 0.19539060 | 9.580764 | 9.632973e-22 | 8.411726e-19 |
| CNAG_00399 | 213.68536 | -2.146601 | 0.22469243 | -9.553507 | 1.253786e-21 | 9.853503e-19 |
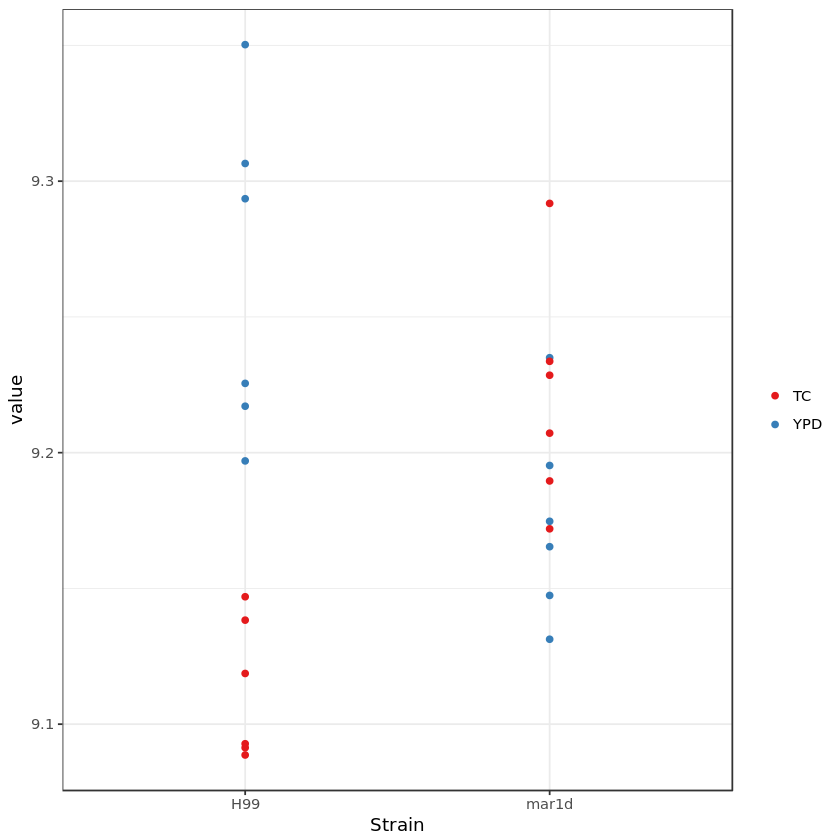
Gene expression value in different media and strain¶
In [11]:
### A function for exploring interactions
myinteractplot <- function(mydds, geneid) {
# plot gene expression value in different media and strain
#
# Args:
# mydds (DESeqTransform object): your input DESeq object
# geneid (Charater): the second count data
#
# Returns:
# (ggplot object) the final plot
assay(mydds) %>%
as_tibble(rownames = "gene") %>%
filter(gene == geneid) %>%
gather(Label, value, -gene) %>%
select(-gene) ->
genedat
colData(mydds) %>%
as.data.frame %>%
as_tibble %>%
full_join(genedat, by="Label") -> genedat
mygeom <- geom_point()
mypal <- scale_colour_manual(name = "", values = brewer.pal(3, "Set1"))
mytheme <- theme_bw()
ggplot(genedat, aes(x = Strain, y = value, color = Media)) + mygeom + mytheme + mypal
}
myinteractplot(rld, "CNAG_05845")
Data type cannot be displayed:
Dendrogram of Samples in Hierarchical Clustering¶
Create Dendrogram of samples using hierarchical clustering
In [12]:
assay(rld) %>%
t() %>%
dist %>%
hclust(method = "complete") %>%
as.dendrogram ->
mydend
Customize function to plot dendrogram showing the sample annotations
In [13]:
dendplot <- function(mydend, columndata, labvar, colvar, pchvar) {
# plot dendrogram
#
# Args:
# mydend (Dendrogram): your input DESeq object
# columndata (DataFrame): the second count data
# labvar (Character): variable that show in label
# colvar (Character): variable that define color
# pchvar (Character): variable that define shape of points
#
# Returns:
# (Dendrogram) final plot of dendrogram
cols <- factor(columndata[[colvar]][order.dendrogram(mydend)])
collab <- brewer.pal(max(3, nlevels(cols)), "Set1")[cols]
pchs <- factor(columndata[[pchvar]][order.dendrogram(mydend)])
pchlab <- seq_len(nlevels(pchs))[pchs]
lablab <- columndata[[labvar]][order.dendrogram(mydend)]
mydend %>%
set("labels_cex", 1) %>%
set("labels_col", collab) %>%
set("leaves_pch", pchlab) %>%
set("labels", lablab)
}
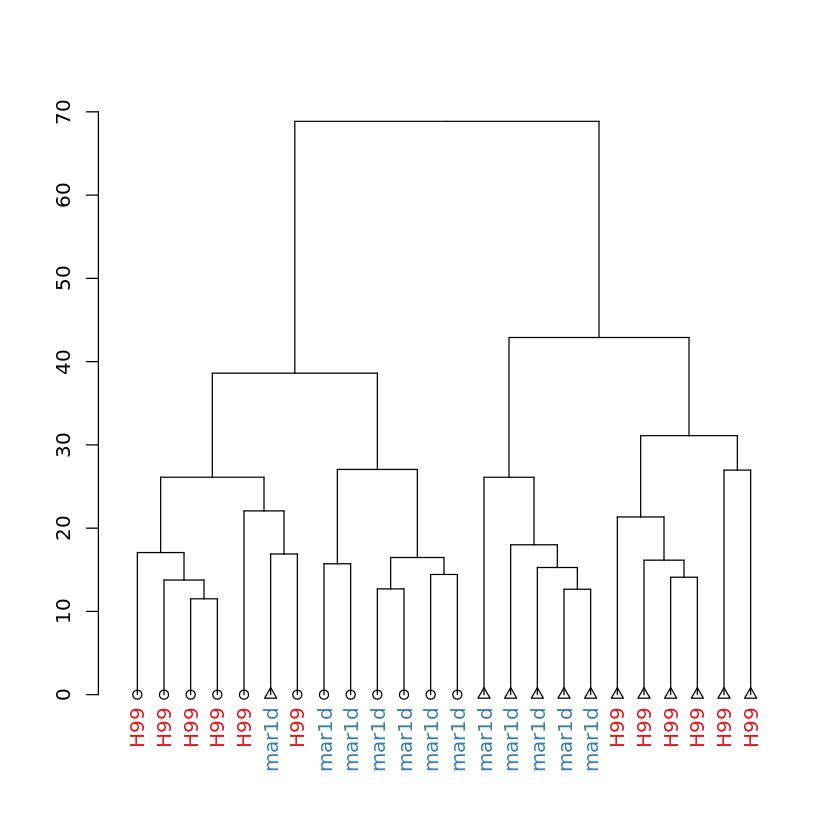
Dendrogram of samples: showing strain of each sample¶
In [14]:
dendplot(mydend, columnData, "Strain", "Strain", "Media") %>% plot
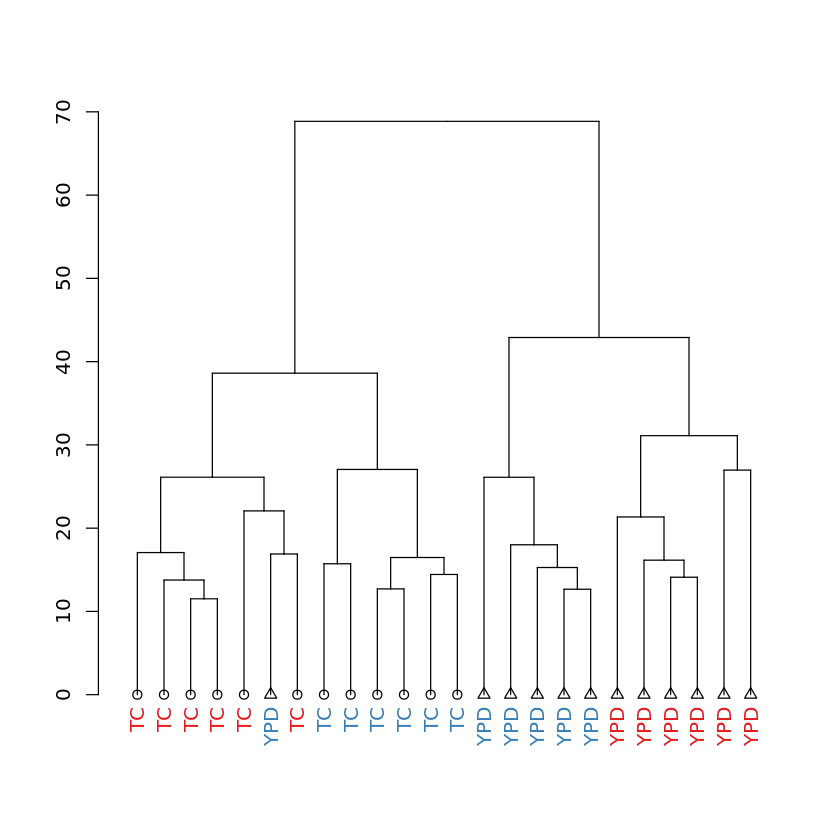
Dendrogram of samples: showing media of each sample¶
In [15]:
dendplot(mydend, columnData, "Media", "Strain", "Media") %>% plot
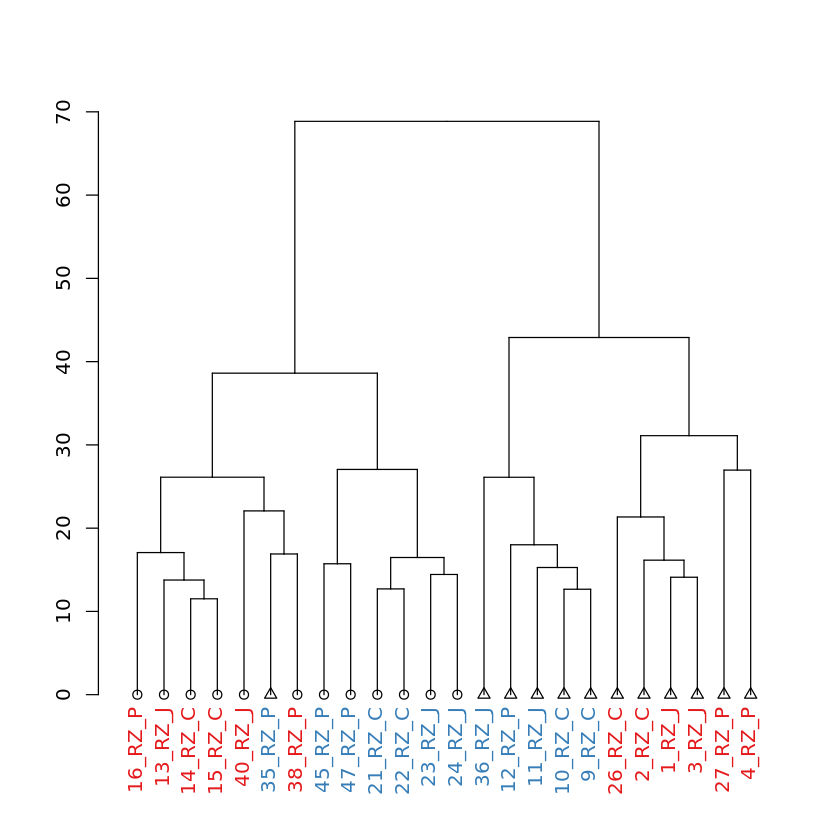
Dendrogram of samples: showing sample names of each sample¶
In [16]:
dendplot(mydend, columnData, "Label", "Strain", "Media") %>% plot
Store the plot into a pdf
In [17]:
pdf(file.path(imgdir, "dendrogram.pdf"))
dendplot(mydend, columnData, "Strain", "Strain", "Media") %>% plot
dendplot(mydend, columnData, "Media", "Strain", "Media") %>% plot
dendplot(mydend, columnData, "Label", "Strain", "Media") %>% plot
graphics.off()